P2-UE10-Zunic-Sémiologie-de-la-polyglobulie_PDF

UE10 – Tissu sanguin
Dr. Zunic
Date : 10/04/2016 Plage horaire : 10h45-12h45
Promo : P2 2016/2017 Enseignant : Dr. Zunic
Ronéistes : Manu SALAUN-PENQUER
Bharath APPAVOUPOULLE
Sémiologie et exploration : polyglobulie, hyperleucocytose et
thrombocytose
I. Polyglobulie
1. Diagnostic positif de la polyglobulie
A. Signes Cliniques
B. Diagnostic Biologique
a) NFS
b) Masse Sanguine
c) Recherche de la mutation de Jak2 V617F
d) Anomalies biologiques à forte valeur diagnostic
e) Autres Examens Biologiques
2. Diagnostic différentiel de la polyglobulie
A. Fausses polyglobulies
B. Polyglobulies secondaires
C. Erythrose pure
3. Evolution
A) Risques liés à la polyglobulie
B) Complications Hématologiques
II. Syndromes Hyperleucocytaires
1. Polynucléose neutrophile
2. Myélémie
III. Sémiologie et Exploration d’une Thrombocytose
1

On va traiter aujourd’hui les syndromes myéloprolifératifs, qui comme ce nom l’indique sont
l’ensemble des symptômes en liant avec une prolifération de la moelle osseuse.
On se souvient du tableau de l’érythropoïèse et de l’hématopoïèse plus généralement (plus loin
dans ce cours également).
On sait qu’il y a une cellule souche (cellule souche hématopoïétique CSH) qui va donner naissance
à l’ensemble des lignées sanguine.
I. Polyglobulie
Nb: le traitement qui date de plusieurs siècles et qui est encore utilisé aujourd’hui, consiste en la
pratique de saignées.
Définition: la polyglobulie (« poly » pour plusieurs et « globulie » pour GR) est une augmentation
de la valeur absolue du nombre d’érythrocyte (Globule Rouge) circulant dans le sang.
!
Sur les hémogrammes, il faut toujours résonner avec les valeurs absolues pour pouvoir poser un
diagnostic, partir sur des démarches diagnostiques et des orientations étiologiques.
• Valeurs normales :
-Hématies .......................... 4,5 - 5,8 (T/L)
-Hémoglobine…………… 13,0 - 17,0 (g/dL)
-Hématocrite ……………. 40,0 – 50,0 (%)
-VGM…………………… 80,0 - 100,00 (fL)
-TCMH .............................. 27,0 - 32,0 (pg)
-CCMH .............................. 32,0 - 36,0 (g/dL)
Nb:
-ce sont le taux d’hémoglobine et l’hématocrite qui vont permettre de définir une polyglobulie.
-VGM = Volume Globulaire Moyen
-TCMH = Teneur Corpusculaire Moyenne en Hémoglobine
-CCMH = Concentration Corpusculaire Moyenne en Hémoglobine
Il y a 2 mécanismes distincts :
- La polyglobulie primitive (ou maladie de Vaquez ou Polycythemia Vera « PV ») : il s’agit d’une
anomalie intrinsèque à la moelle osseuse, une anomalie de la cellule souche hématopoïétique qui
va soudainement s’expandre de manière inappropriée (caractéristiques tumorales de prolifération)
entraînant une amplification de la lignée érythroïde. On aboutit ainsi à une anomalie du nombre
de GR.
Les précurseurs érythroblastiques sont anormaux avec une sensibilité accrue à l’hormone de
stimulation de l’érythropoïèse, l’érythropoïétine.
- La polyglobulie secondaire : la moelle osseuse est normale et elle va répondre à des stimuli
extérieurs (extra-hématologique) qui vont conduire à une augmentation des GR, il s’agit d’une
réponse appopriée de l’érythropoïèse.
2

Exemple: une des causes de polyglobulie secondaire, à rechercher lorsqu’on nous adresse un
patient pour une polyglobulie:
Le principal stimulus de l’érythropoïétine étant la pression en O2 dans le sang, chez un patient en
hypoxie chronique (insuffisant respiratoire), la sécrétion / la fabrication de l’érythropoïétine va être
augmenté, et donc la moelle osseuse va recevoir comme message de fabriquer plus de globules
rouge => on est dans le cas d’une polyglobulie secondaire à une insuffisance respiratoire.
SCHEMA DE L’HÉMATOPOÏÉSE
Pour rappel, c’est une CSH (cellule souche hématopoïétique) qui va donner naissance in fine à
l’érythrocyte ou globule rouge.
On parle de polyglobulie vraie lorsqu’il y a une augmentation du volume globulaire total.
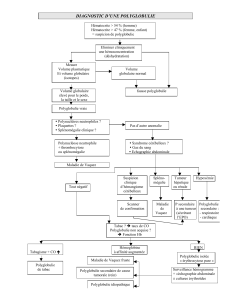
Tout d’abord, on cherche à savoir si on a une polyglobulie vraie ou fausse et pour cela on réalise
un examen de la masse isotopique de la masse sanguine.
Si c’est une polyglobulie vraie on entreprend une démarche diagnostique pour savoir si c’est une
polyglobulie primitive ou secondaire par des examens d’exploration.
Dans le cas d’une polyglobulie primitive, il s’agit d’une maladie clonale c’est-à- dire qu’il y a une
anomalie génétique en l’occurence ici une mutation du gène d’une kinase qu’on appelle la JAK2
qui est une enzyme qui fait partie du récepteur de l’érythropoïétine au niveau de la lignée
érythroïde. Cette mutation va donner une hypersensibilité de la lignée érythroblastique à
l’érythropoïétine ce qui explique le nombre accru de GR. On retrouve cette mutation de manière
très majoritaire dans la polyglobulie primitive mais également dans d’autres syndromes
myéloprolifératifs.
3

1. Diagnostic positif de la polyglobulie
!
A. Signes Cliniques
Epidémiologie: - personnes âgées de plus de 50 ans (le plus souvent)
- touche davantage des hommes avec un sex ratio de 1,2!
Nb: un jeune homme sportif avec un légère excès de globules rouge, c’est quasi certainement une
polyglobulie secondaire.
Signes non spécifiques de la polyglobulie:
-Érythrose : coloration rouge pourpre, constante prédominant sur la zone palmo-faciale.
-Syndrome d’hyperviscosité sanguine : qui se traduit avant l’ischémie et la nécrose par des
signes qu’il faut chercher à l’interrogatoire :
• Céphalées (pulsatiles en étau et permanente, prenant toute la boîte crânienne et qui ne sont
calmées par aucun traitement antalgique ou position puisqu’elles sont dûes à l’oblitération
capillaire au niveau cérébral), vertiges.
• Acouphène (bourdonnement d’oreille), scotomes (petites tâches blanches qui apparaissent
en raison des microthrombi de l’artère centrale de la rétine lorsque l’hyperviscosité du sang
est important, lors d’un fond d’oeil on aperçoit des petites hémorragies en flamèche puisque
les capillaires bouchés vont s’ouvrir et laisser passer des petites hémorragies)
• Erythromélalgies (rougeurs douloureuses des extrémités [jambes, mains, visages] qui sont le
signes de mauvaise circulation [pas ischémie mais presque] et qui peuvent être très
invalidantes pour le patient). !
Nb: On valide le diagnostic par une prise de sang.
-Complications révélatrices :
• Troubles ischémiques: des vaisseaux de gros calibre en particulier les AVC qui vont se
révéler par des signes neurologiques déficitaires notamment une aphasie, une hémiparésie
ou une hémiplégie.
• Manifestations hémorragiques: en aval un capillaire bouché sous la pression va “exploser” et
entraîner une hémorragie => donc les patients avec une polyglobulie primitive, on a souvent
un épistaxis, aussi hémorragies dans l’oeil etc.!
-Signes cliniques évocateurs de la maladie de Vaquez:
• Un prurit intense exacerbé par l’eau (donc après la douche)
• Splénomégalie modérée (entre 12-15 cm lors de la palpation en inspiration profonde) dans
75% des cas. !
4

B. Diagnostic Biologique
a) NFS
*Anomalie de la lignée rouge: non spécifiques
-une augmentation du nombre des GR
-une augmentation du taux d’Hb : >17 g/dL chez l’homme et > 16g/dL chez la femme
-pas d’anomalie de morphologie des GR
-réticulocytes normaux (forme normale) [elle dit également “taux de réticulocytes normal“]
*Augmentation d’une des 2 autres lignées: important à regarder sur un hémogramme
-hyperleucocytose (50% des cas)
•Par polynucléose (10000 à 30000 /mm3)
•Basophilie, éosinophilie
•Myélémie discrète (passage des cellules de la moelle osseuse dans le sang (5% des éléments)
-hyperplaquettose (75% des cas) qui peut être relativement importante à plus de 1000000/mm3
dans 10% des cas.
Nb: Très simplement, avec un hémogramme on peut déjà bien avancer sur le diagnostique de
polyglobulie et avoir déjà des orientations étiologiques.
!
b) Masse Sanguine
L’étude isotopique de la masse sanguine:
-Seule technique permettant d’affirmer que c’est une vraie polyglobulie.
-Inutile si le taux d’Ht >60% => on n’a pas de fausse polyglobulie à des taux aussi élevé.!
Dans cet examen: une augmentation du volume globulaire total (VGT):
->20% de la valeur théorique calculée
-En valeur absolue : >36mL/kg chez l’homme; >32 mL/kg chez la femme
=> ceci est la définition stringente de la polyglobulie;
Cet examen élimine les fausses polyglobulies
Lorsqu’on fait la masse sanguine, on calcule le
volume plasmatique (en jaune), et le volume
globulaire (en rouge). Les cellules les plus
nombreuses sont les globules rouge donc le
volume globulaire correspond de manière assez
corrélé à la masse globulaire total.
- Normal: répartition classique;
- Vraies Polyglobulies: (moelle osseuse produit
un excès de globule rouge), on a une masse
globulaire totale qui est augmenté au dépend du
volume plasmatique. Tout les valeurs analysés
sur l’hémogramme et à l’étude isotopique de la
masse sanguine sont augmentés.
5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
1
/
23
100%