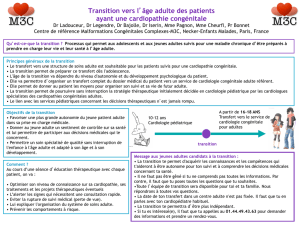
Cardiopathies congénitales et pédiatriques

8/01/13
Cardiopathiescongénitalesetpédiatriques
Les malformations cardiaques congénitales sont fréquentes et souvent graves.
Savoir les comprendre et les traiter est une spécialité à part entière. Un enfant sur
100 naît avec une cardiopathie congénitale (CC) ; un tiers d’entre eux aura besoin
d’un traitement par cathétérisme interventionnel ou chirurgie cardiaque ; au moins un
tiers de ceux-ci aura besoin de nouvelles interventions lourdes à l'âge adulte. Ceci
représente pour l’instant un volume d’activité d’environ 4000 interventions
chirurgicales par an, et autant de cathétérismes cardiaques, presque tous
interventionnels (thérapeutiques) qui doivent, pour avoir de bons résultats, se faire
avec un très haut niveau d'exigence et d'expérience, maintenu par une forte activité.
Tous ces enfants auront besoin d’un suivi spécialisé qui se prolonge toute la vie.
Aujourd’hui, plus de 85% des patients survivent à leur cardiopathie et donc atteignent
l’âge adulte. En France, on estime que la population de patients ayant une
cardiopathie congénitale est de plus de 300 000 individus avec autant d’enfants que
d’adultes. Les mêmes équipes prennent aussi en charge les cardiopathies acquises
de l’enfant.
La filière de soins est particulièrement fragile, mal définie, cherchant sans vraiment la
trouver, une place équilibrée entre les spécialités pédiatriques et adultes, entre le
public et le privé, entre les schémas types maladies rares et la masse de patients à
prendre en charge.
La population de cardiaques congénitaux adultes croît ; c’est la rançon du succès
des traitements appliqués aux enfants. Avec cette croissance, un certain nombre de
problématiques spécifiques émergent sur la continuité des soins, l’organisation des
équipes et des structures, la formation des médecins et l’information-éducation des
patients. On anticipe également que l’état sanitaire de cette population va se modifier
avec l’émergence de complications attendues ou pas. En effet, cette population est
totalement nouvelle, les personnes concernées, adultes, étant les premiers humains
ayant survécu à une cardiopathie congénitale auparavant létale.
La charge de travail va donc croître de façon certaine. Les modalités de ce travail
doivent aussi s’adapter et évoluer en conséquence.
La démarche de ce document est de jeter les bases d’un processus d'analyse de la
situation actuelle et d’anticipation de ces problèmes à venir pour que cette activité de
soin se fasse de façon sûre et pérenne sur l’ensemble du territoire, avec la même
qualité.

8/01/13
Epidémiologierécente
Cette filière spécialisée prend en charge d’une part les patients, du fœtus à l’adulte,
ayant des CC, et d’autre part, les enfants qui acquièrent une cardiopathie.
Prévalencedescardiopathiescongénitalestendances
Plusieurs études françaises [1],[2] et européennes[3] donnent une prévalence néonatale
des CC d’un peu moins de 1%, soit pour la France, environ 6 à 8000 nouveau-nés
par an.
Le diagnostic est fait chez le fœtus pour environ la moitié des CC et pour près de
80% des formes complexes. Le dépistage puis le suivi prénatal de ces CC fœtales
représente donc une part importante de l’activité. Les couples/mères suivent alors un
parcours pluridisciplinaire à la fois précis et complexe au sein des structures de prise
en charge périnatale des CC.
Quand il n’est pas fait chez le fœtus, le diagnostic peut être fait en période néonatale
ou dans l’enfance. Il reste que certaines malformations ne sont vues qu’à l’âge adulte
car elles sont peu voire pas symptomatiques avant qu’elles ne se compliquent après
plusieurs décennies.
En 2007, la province du Québec a fait un recensement exhaustif des patients ayant
des CC à partir de deux bases de données, celle de la Régie de l’Assurance Maladie
du Québec, de 1983 à 2000 et celle de la MedEcho de 1987 à 2000[4]. Cette étude a
permis d’avoir une estimation de la prévalence des CC à l’âge adulte et de connaître
son évolution ces trois dernières décennies (figure 1).
De 1985 à 2000, la prévalence des CC au Québec est passée de 6,88 à 11,89/1000
enfants et de 3,57 à 4,09/1000 adultes. La prévalence des CC était donc de
5,78/1000 pour l’ensemble de la population.
Au cours de cette même période, l’âge médian des patients est passé de 11 ans
(IQR, 4 à 22 ans) à 17 ans (IQR, 10 à 28 ans) (p<0,0001). En 2000, l’âge médian
des adultes ayant une CC était de 40 ans (IQR, 22 à 39 ans) et ils représentaient
55% de la population totale des CC. Mieux, le taux de CC sévères chez l’adulte avait
augmenté en proportion d’environ un tiers à presque la moitié de la population totale
des CC de l’adulte (figure 2).
Figure 1. Evolution des prévalences des CC (CHD) chez l’enfant (<18 ans) et l’adulte
entre 1985 et 2000

8/01/13
Figure 2.
De la même façon, la prévalence des CC sévères de l’adulte a augmenté de 85% en
15 ans (0,21 à 0,38/1000 adultes) alors que dans le même temps cette augmentation
était nettement plus faible chez l’enfant d’environ 22% (1,15 à 1,30/1000 enfants).
Très clairement, ces augmentations de prévalence sont liées à une augmentation
considérable de la survie à tous les âges en particulier pour les formes complexes
[5],[6]. Les progrès faits dans le domaine des soins médicaux et chirurgicaux pour tous
les types en sont la cause directe. L’impact du diagnostic prénatal sur le nombre de
naissances vivantes d’enfants ayant une CC est resté très stable depuis plus de 20
ans. Ceci a pour conséquence une stabilité du nombre d’enfants à prendre en
charge pour une CC. En extrapolant les chiffres de leur étude, les auteurs estiment
que, au minimum, 181 000 Canadiens et 1,8 million d'Américains étaient atteints de
CC en 2000, soit un enfant sur 85 et un adulte sur 250. Il est possible avec les
mêmes méthodes d’estimer le nombre de CC pour plusieurs pays. Ainsi il y avait, en
2000, au minimum 180 000 patients porteurs de CC en France :
Population CC
sévère CC
modérée CC simple Totale
CC
France 64 000 000 26 676 68 856 83 904 179 618
Allemagne 82 000 000 34 164 88 184 107 456 230 037
Italie 58 000 000 23 400 81 540 99 360 163 074
Belgique 10 000 000 4 178 11 174 13 616 29 119
Pays Bas 16 000 000 6 669 17 214 20 976 44 904
Grèce 11 000 000 4 596 12 291 14 977 32 030
Prévalence brute des cardiopathies congénitales
Incidencedescardiopathiescongénitales–Diagnosticprénatal
D’après l’étude européenne EUROCAT [7], la prévalence des CC en Europe est de
8/1000 et inclue 89,7% de naissances vivantes, 1,6% de décès périnataux (20

8/01/13
semaines à 8 jours de vie) et 8,7% d’interruptions de grossesse. Si les CC associées
aux anomalies chromosomiques étaient exclues (essentiellement des trisomies 21),
la prévalence totale serait de 7 pour 1 000 avec une grande variabilité entre les pays.
EUROCAT estime que, sur 3,3 millions de naissances annuelles dans l’Union
Européenne, environ 36 000 enfants naissent avec une CC, que celles-ci sont la
cause de 1250 décès périnataux et que le diagnostic prénatal des CC sans
anomalies chromosomiques conduit à 2000 interruptions de grossesse.[8] En France,
les pourcentages d'interruption de grossesse pour CC sont parmi les plus élevés
(1,34/1000 naissances) pour anomalies cardiaques non chromosomiques. Ils sont
stables depuis plus de 20 ans (données EpiCard).
Il est important de noter qu’il n’y a pas de différence régionale de l’incidence par
patients domiciliés. En théorie, la pratique du transfert in utero peut modifier
l’incidence régionale à cause de transferts vers les centres experts. La prévalence
des CC reste cependant mal connue en France. Il est possible qu’elle ne soit pas
homogène car parfois les patients les plus sévères finissent par vivre dans la région
du centre expert qui les prend en charge.
Augmentationdel’incidencedesadultescardiaquescongénitauxenFrance
Le nombre de patients adultes ayant des cardiopathies congénitales se situe entre
2000 et 3000 patients par million d’habitants et la moitié d’entre eux a une CC
suffisamment sévère pour nécessiter une prise en charge médicale et/ou
chirurgicale. On peut donc estimer, qu’en France, au moins 100 000 à 150 000
patients sont concernés et que 50 000 à 75 000 doivent être pris en charge de façon
chronique. Cette population augmente régulièrement et très rapidement depuis ces
dernières années grâce aux progrès considérables de la médecine et de la chirurgie
cardiaques pédiatriques depuis une quinzaine d'année; ainsi, la plupart des 3000 à
4000 jeunes enfants opérés maintenant chaque année vont survivre jusqu’à l’âge
adulte [9].
L’émergencedescomplicationscardiaqueschezlescongénitauxadultes
Le développement récent et rapide de cette population d’adultes cardiaques
congénitaux est accompagné par la mise en évidence des complications spécifiques
à leur état, dont certaines sont encore à mieux décrire. Schématiquement, on peut
les différencier entre :
- les complications qui sont en lien direct avec la malformation : elles sont
multiples, et plus ou moins caractéristiques. Ainsi, un adulte opéré d’une
tétralogie de Fallot dans l’enfance est typiquement totalement
asymptomatique jusqu’à 25 à 30 ans. Ensuite vont apparaître des troubles liés
à l’insuffisance valvulaire pulmonaire (insuffisance cardiaque droite), des
troubles du rythme ventriculaires, puis plus tard éventuellement une dilatation
de l’aorte ascendante.
- les complications qui proviennent de la coexistence de la pathologie
congénitale et d’autres états particuliers comme la grossesse ou maladies
comme une endocardite, un cancer, une autre chirurgie ou même une grippe.
- les complications sociétales et professionnelles. Reliées à la pathologie et/ou
son traitement, elles représentent l’ensemble des troubles d’acquisition,
d’insertion et de maintien dans la “normalité“ de l’emploi, des capacités

8/01/13
fonctionnelles et des structures familiales. On peut y relier des troubles
psychologiques voire psychiatriques.
Lescardiopathiesacquisespédiatriques
La filière de cardiologie et de chirurgie cardiaque pédiatrique prend également en
charge les enfants qui souffrent de cardiopathies non malformative ou acquises. Les
principales sont les suivantes :
Troubles du rythme cardiaque comprenant aussi les maladies héréditaires du rythme
et de la conduction et les troubles du rythme compliquant une cardiopathie
congénitale opérée. Cette activité représente environ 50% de l’activité de cardiologie
congénitale adulte.
Valvulopathies du rhumatisme articulaire aigu à prévalence très faible, mais restant
très fréquente dans les régions d’origine de l’immigration vers la France et dans les
régions d’outre mer.
Endocardite infectieuse dont l’incidence ne baisse pas et qui survient volontiers sur
cardiopathie congénitale.
Cardiomyopathies rares mais très consommatrices de ressources et principales
indications de transplantation cardiaque.
L'hypertension pulmonaire rare dans sa forme idiopathique mais représentant 5% de
l’activité de cardiologie pédiatrique quand elle complique d'autres pathologies non
cardiaques.
Enfin, il y un une variété importante de maladies rares liées à d'autres problèmes
pédiatriques comme les coronaropathies de l'enfant liée à la maladie de Kawasaki ou
à une hypercholestérolémie familiale, les cardiopathies des erreurs innées du
métabolisme, les tumeurs cardiaques, les cardiomyopathies liées aux traitements
anticancéreux des enfants, etc.
La somme de ces constats (augmentation du dépistage prénatal des CC, stabilité
des naissances vivantes et augmentation considérable de la survie) conduit à une
augmentation de la charge de travail liée à l’augmentation de la prévalence des CC
en population.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
1
/
30
100%