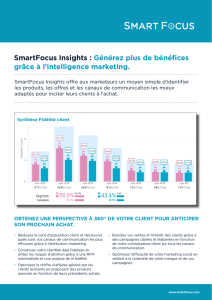
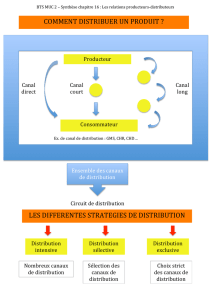
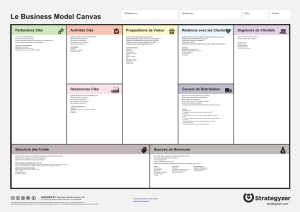
thèse - Université Francois Rabelais

UNIVERSITÉ FRANÇOIS - RABELAIS
DE TOURS
ÉCOLE DOCTORALE "Santé, Sciences, Technologies"
UNITE INSERM U921 "NUTRITION CROISSANCE ET CANCER"
THÈSE
présentée par :
Ludovic GILLET
soutenue le : 3 septembre 2010
pour obtenir le grade de : Docteur de l’université François - Rabelais
Discipline/ Spécialité : Sciences de la Vie et de la Santé
IMPLICATION DES CANAUX SODIQUES
DÉPENDANTS DU VOLTAGE DANS
L'INVASIVITÉ DE CELLULES CANCÉREUSES
MAMMAIRES ET RÉGULATION PAR L'ACIDE
DOCOSAHEXAÈNOÏQUE
THÈSE dirigée par M. LE GUENNEC Jean-Yves, Professeur des Universités, Université Montpellier 2
et co-encadrée par M. BESSON Pierre, Maître de Conférences, Université de Tours
RAPPORTEURS :
Mme LAGADIC-GOSSMANN Dominique Directeur de Recherche CNRS, Rennes
M. SORIANI Olivier Maître de Conférences, HDR, Université de Nice
JURY :
M. BESSON Pierre Maître de Conférences, Université de Tours
M. BOUGNOUX Philippe Professeur des Universités, Université de Tours
M. KELLENBERGER Stephan Maître d'Enseignement et de Recherche, Université de Lausanne
Mme. LAGADIC-GOSSMANN Dominique Directeur de Recherche CNRS, Rennes
M. LE GUENNEC Jean-Yves Professeur des Universités, Université Montpellier 2
M. ROGER Sébastien Maître de Conférences, Université de Tours
M. SORIANI Olivier Maître de Conférences, Université de Nice
M. VAN COPPENOLLE Fabien Professeur des Universités, Université Lyon 1
1

2

Ce travail de thèse a été réalisé dans le laboratoire "Nutrition, Croissance et Cancer",
Unité INSERM U921, Université François-Rabelais de Tours,
Dirigé par le Pr. Philippe Bougnoux.
Cette thèse a été financée par le Conseil Régional de la Région Centre pendant les trois
premières années. La quatrième année de thèse a été effectuée dans le cadre d'un contrat
d'attaché temporaire d'enseignement et de recherche à temps partiel (A.T.E.R Doctorants).
Thèse dirigée par le Pr. Jean-Yves Le Guennec
et co-encadrée par le Dr. Pierre Besson.
3

4

Remerciements
Après ces quatre merveilleuses années passées au laboratoire (voire ces dix très belles
années passées à l’université), je ne suis pas certain que de simples mots suffiront à vous dire
merci comme vous le méritez…
Je tiens à remercier tout d'abord le Pr. Philippe BOUGNOUX. Merci de m’avoir
accueilli dans votre laboratoire et d’avoir accepté de faire partie de mon jury de thèse. Merci
également pour toutes vos réflexions, analyses ainsi que pour les conseils que vous avez pu
me prodiguer, que cela concerne la thèse ou bien l’après thèse…
Je voudrais remercier tout particulièrement Jean-Yves LE GUENNEC et Pierre
BESSON pour leur excellent encadrement, mais aussi pour tout ce qu’ils m’ont apporté
pendant ces quatre années de thèse.
Jean-Yves, avant même d’être mon directeur de thèse, tu as été mon professeur
d’électrophysiologie, dès la licence. La qualité de tes cours ainsi que le professeur sortant un
peu de l’ordinaire que tu étais m’ont beaucoup aidé dans le choix de la voie que je suis
aujourd’hui. Merci d’être toujours à l’écoute des étudiants et de nous guider lorsque nous en
avons besoin. Il est vrai qu’en licence je n’aurais jamais imaginé faire une thèse avec toi ; je te
remercie de m’avoir donné cette chance. Merci pour tous tes conseils que ce soit avant ou
pendant la thèse. Je me répète mais merci encore pour ton encadrement, merci pour ta
confiance et surtout merci de m’avoir aidé à développer mon esprit critique. Merci également
pour tous les bons moments passés avec toi pendant ces quelques (trop courtes) années de
thèses. Merci de m’avoir permis de participer à de nombreux congrès scientifiques, et par la
même occasion de découvrir les restaurants Tex-mex du monde entier (en particulier celui de
Genève…). Je te remercie également de m’avoir donné l’envie de renouer avec le monde des
arts martiaux pendant ma thèse, et de m’avoir fait découvrir par ailleurs de nouveaux horizons
musicaux (aaaah Higelin…). Enfin, même si tu as dû t’éloigner pendant ma dernière année de
thèse, au final tu n’étais jamais très loin…
Pierre, merci à toi d’avoir été toujours présent pour répondre, quelle que soit l’heure
de la journée ou de la soirée, aux moindres questions que je me posais. Merci de nous faire
partager au quotidien tes innombrables connaissances et d’avoir à chaque fois la solution à
nos problèmes. Ce fût une réelle chance et un très grand plaisir que de t’avoir comme
encadrant de thèse. Tu es je pense la personne la plus cultivée que je connaisse, mais
également la plus modeste… Merci de m’avoir transmis une certaine rigueur scientifique,
mais aussi tous tes petits trucs et astuces utiles au quotidien… Je te remercie également pour
tous les bons moments passés en ta compagnie que ce soit au labo, en congrès ou au cours des
différentes soirées…
5
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
1
/
240
100%