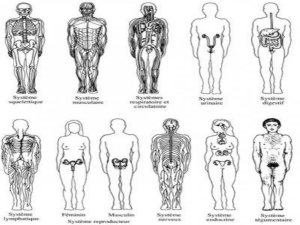
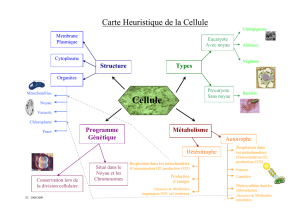
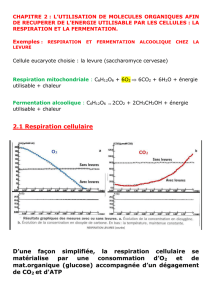
Lire l`article complet

La mitochondrie :
une histoire récente
pour une vieille relique
Mitochondria: a recent story for an old relic
■
■
P. Bouche*
L’existence des mitochondries semble remonter à fort loin. En effet, les mitochondries
sont des restes d’intrus indépendants, de nature bactérienne, qui ont pris place de façon
permanente dans nos cellules il y a quelque billions d’années. C’est sans doute pour
cette raison que les mitochondries possèdent toujours leur propre ADN circulaire qui code
pour 13 des 90 protéines du travail terminal du métabolisme mitochondrial. Si l’existence
de ces mitochondries est fort ancienne, l’histoire de leur pathologie est bien récente. C’est
à Rolf Luft, un endocrinologue de l’université Karolinska de Stockholm, que revient le
mérite d’avoir rapporté le premier cas, en 1962, d’une mitochondriopathie chez une jeune
femme qui présentait un état d’hypermétabolisme sévère sans hyperthyroïdie. Une
anomalie mitochondriale fut mise en évidence par des études ultrastructurales du
muscle et par des anomalies biochimiques du couple oxydation/phosphorylation. Il est
assez paradoxal de constater que le premier cas de mitochondriopathie concerne une
pathologie rarissime. Quelques années plus tard, deux neurologues de l’université de
Pennsylvanie (Shy et Gonatas, 1964 et 1966) ont rapporté des anomalies mitochondriales
musculaires chez des enfants atteints de myopathie ; ils leur donnèrent des noms grecs
quelque peu fantaisistes : myopathie pléoconiale (nombreuses mitochondries anormales)
et myopathie mégaconiale (mitochondries géantes). Ces auteurs avaient alors émis
l’hypothèse que ces deux types de myopathie pouvaient être dus à une anomalie
génétique de l’ADN mitochondrial. King Engel a introduit une technique histochimique
simple qui a fait date pour détecter les proliférations mitochondriales du muscle, par
une coloration modifiée du trichrome de Gomori. Ces fibres musculaires anormalement
colorées furent alors nommées ragged-red fibers, et leur présence deviendra la “marque
de fabrique” de la myopathie mitochondriale, indispensable au diagnostic. De nombreux
syndromes seront décrits par la suite, dont le point commun est l’anomalie
mitochondriale. Les plus fréquents sont l’ophtalmoplégie externe progressive isolée ou
associée à d’autres atteintes comme le syndrome de Kearns-Sayre, les syndromes MERFF
et MELAS, et le syndrome de Leigh, qui associe une encéphalopathie aux autres atteintes.
Les spécialistes mondiaux, que l’on nomme volontiers les “mitochondriaques”,
ont ensuite relevé les différents déficits biochimiques de la chaîne respiratoire. Puis vint
1988, ère de la génétique moléculaire mitochondriale, qui pendant la décennie suivante
va explorer les différentes délétion de l’ADN mitochondrial, si bien que plus
d’une centaine de mutations de l’ADN mitochondrial purent être observées en association
avec divers syndromes multisystémiques ou spécifiques d’un tissu, qu’ils soient hérités
de la mère ou sporadiques. C’est ainsi que, en 1998 et 1999, Michio Hirano mit
en évidence une liaison au chromosome 22 et surtout une mutation du gène codant pour
la thymidine phosphorylase (TP) dans le syndrome MNGIE. Une nouvelle ère commençait,
celle des mutations nucléaires de gènes codant pour des protéines mitochondriales,
3
Correspondances en Nerf & Muscle - Vol. III - n° 2 - octobre 2005
éditorial
*Service des explorations fonctionnelles,
hôpital de la Pitié-Salpêtrière, Paris.

4
Correspondances en Nerf & Muscle - Vol. III - n° 2 - octobre 2005
éditorial
annonçant, enfin, l’arrivée du “périphérologue”. En effet, les mitochondriopathies
ne semblaient intéresser le nerf périphérique que de façon contingente, histoire de dire
que les syndromes étaient multisystémiques. Avec le MNGIE déjà, la neuropathie
périphérique faisait partie intégrante du syndrome et parfois, certes très rarement,
au premier plan. C’est surtout avec le syndrome SANDO et le CMT2A que les choses vont
radicalement changer. Le syndrome SANDO associe une neuropathie ataxiante,
une dysarthrie et une ophtalmoplégie. La fréquence de tels cas est, apparemment,
actuellement rare, mais sans doute sous-estimée. L’ophtalmoplégie et la dysarthrie
peuvent manquer ou être au second plan. Il faut maintenant penser à ce diagnostic
devant une neuropathie ataxiante chronique. Pour le deuxième syndrome,
c’est en quelque sorte la démarche inverse qui s’est produite. Le CMT axonal de type 2
était connu et, à ce jour, seule une famille présentait une mutation génétique.
C’est en reprenant tous les autres cas de CMT2A, lié au chromosome 1, que l’on a pu
mettre en évidence une mutation de la mitofusine 2 (MFN2) de l’ADN nucléaire.
On se trouve donc en présence de deux syndromes périphériques opposés : l’un, sensitif,
qui correspond plutôt à une ganglionopathie et l’autre moteur, s’apparentant
à une neuropathie axonale. Dans le premier cas, le gène responsable le plus souvent
intéressé est la POLG (polymérase gamma). L’ophtalmoplégie externe progressive
à hérédité mendélienne, récessive ou dominante, est le plus souvent due à une mutation
POLG. La polymérase gamma est un enzyme dont l’action est assez complexe et,
chez la souris déficitaire en POLG, présente un tableau typique de souris âgée,
ce qui semble impliquer les mitochondries dans le vieillissement. Il est donc intéressant
de noter que l’altération d’un enzyme impliqué dans le vieillissement est susceptible
de provoquer une ganglionopathie. En fait, le mécanisme responsable est
vraisemblablement plus complexe, mais il faut retenir de cela qu’une ganglionopathie
presque pure, d’allure chronique, peut être provoquée par une altération mitochondriale.
Dans le deuxième syndrome CMT2A, c’est l’altération d’une protéine mitochondriale
de fusion, la MFN2 qui est impliquée dans la genèse de la neuropathie. Un modèle
animal, la souris knockout hétérozygote MFN2, présente un phénotype normal,
mais une altération mitochondriale. La mobilité et le transport des mitochondries sont
des éléments clés pour le fonctionnement axonal, notamment du nerf périphérique.
La mutation de la MFN2 est la cause principale du CMT2A et cette découverte ouvre
de très intéressantes perspectives dans la compréhension de la physiopathologie
des polyneuropathies axonales, acquises et héréditaires. De futures études viendront
sûrement éclairer et alimenter ce nouveau chapitre de la pathologie du nerf
périphérique.
De cet éditorial, qui est plutôt une revue historique mettant en perspective le rôle des
mitochondries dans la pathologie nerveuse périphérique, il ressort qu’après l’ère
immunologique, une nouvelle ère débute, celle de la mitochondrie. Il n’est pas impossible
que des altérations mitochondriales soient également responsables d’autres pathologies
comme les maladies du motoneurone... Affaire à suivre ! ■
1
/
2
100%