05-1 - David - INRA Versailles

BIOFUTUR 266 • MAI 200622
Dossier
Génome et diversité ches les plantes cultivées
L
a diversité des êtres vivants est spectaculaire. Nos
repères naturels de perception de la diversité nous
permettent d'apprécier l'étendue de l'adaptation
des différentes formes, innombrables et entremêlées.
La biodiversité s'exprime en particulier sous la forme
d’espèces différentes, et d'individus différents au sein de
la même espèce. Les observations directes, plus ou moins
élaborées, de plantes nous révèlent des palettes de formes,
de couleur, de taille, de vitesse de développement, de résis-
tance aux maladies ou de réactions à d'autres facteurs
environnementaux, qui sont autant de phénotypes.
P
our les plantes sauvages, cette diversité est souvent
interprétée comme le résultat d’une sélection pour
l’adaptation à des conditions de vie ou de milieu
contrastées. Par exemple, la production de graines
d’une plante dépend de sa date de floraison. Ainsi, dans
un milieu stressant, des plantes précoces pourront avoir
plus de descendants que des plantes tardives (on parle
d’une meilleure valeur sélective). Les plantes précoces
y seront donc plus fréquemment rencontrées tandis
que des plantes plus tardives s’épanouiront dans les
environnements plus favorables.
P
our autant, ce type d’adaptation est souvent difficile
à mettre en évidence, et il est probable qu’une part
importante de la diversité observée ne joue pas de rôle
majeur dans l’adaptation des plantes. Celle-ci est alors
considérée comme sélectivement neutre et évolue au
gré du hasard. Pour les plantes cultivées, cette diver-
sité prend le nom de ressources génétiques. Elle est uti-
lisée par les améliorateurs des plantes depuis leur
domestication, pour adapter ces dernières aux
contraintes de l’agriculture et à ses évolutions suc-
cessives. Pour les plantes sauvages, elle représente un
réservoir d’innovation et d’adaptation.
Le regard de la génomique
sur la diversité naturelle
des plantes cultivées
Les nouvelles technologies d’étude du génome explorent la diversité des
plantes au niveau même de l’ADN. L’accumulation récente de données
donne accès à de nouvelles connaissances sur l’histoire des plantes mises
en culture par l’homme. L’étude de la diversité des plantes, notamment
au niveau des gènes, permet de tester des théories évolutives sur le rôle
de la sélection pour l’adaptation, avec des retombées qui permettront
de repérer au sein même des gènes les nucléotides impliqués dans la
variation des plantes entières et par-là même de décrypter les facteurs
d’adaptation des plantes à leur environnement, naturel ou anthropique.
Jacques David*, Olivier Loudet**, Jean-Christophe Glaszmann***
* UMR Inra / Ensam /
IRD Diversité et génome
des plantes cultivées, Inra,
Domaine de Melgueil,
34130 Mauguio
** SGAP, Inra,
Route de Saint Cyr,
78026 Versailles cedex
*** Cirad Montpellier,
bât 3, BP 5035,
34032 Montpellier cedex 1
05-1 - David 13/04/06 9:31 Page 22

BIOFUTUR 266 • MAI 2006 23
La diversité trouve sa source dans l’ADN
N
ous disposons d'autres moyens d'appréhender cette
diversité. Au niveau des plantes entières, les différences
visibles se rapportent bien sûr à l’existence de varia-
tions génétiques codées dans l’ADN des individus,
appelées polymorphismes. Ces variations sont créées
par différents types de mutation, et certaines sont sou-
mises à une sélection en rapport avec leur effet sur un
comportement phénotypique.
T
outefois, au niveau de l’ADN, la plupart des varia-
tions observées sont sélectivement neutres et ne confè-
rent a priori aucun avantage sélectif. La génomique
fournit un grand nombre de marqueurs moléculaires,
de différents types, permettant de révéler des poly-
morphismes sur l'ensemble du génome, de façon quasi-
aléatoire aussi bien ciblée sur des gènes « candidats »
ayant des fonctions présumées
(encadré 1)
. Elle produit
donc des outils très puissants pour étudier la diversité
génétique des populations de nombreuses espèces
sauvages et cultivées, en particulier pour en retracer
l’histoire (phylogéographie) et en comprendre la dyna-
mique (colonisation, isolement reproducteur, adap-
tation, déclin et disparition).
P
ar exemple, les phylogénies entre espèces sont éta-
blies par comparaison de séquences de gènes. Elles per-
mettent de vérifier si les taxonomies mises en place par
les botanistes sur des critères floristiques se retrouvent
au niveau du génome. Ce travail donne des indications
sur les proximités génétiques entre espèces, permettant
notamment d’identifier les ancêtres les plus probables
des espèces cultivées, comme la téosinte (Zea diplo-
perennis), espèce allogame
*1
et ancêtre du maïs.
Mieux comprendre l’histoire
et le fonctionnement des populations
E
n premier lieu, la diversité peut être comparée entre
espèces. Ainsi, la téosinte possède une diversité bien
supérieure à celle de l’orge sauvage (Hordeum spon-
taneum), elle-même supérieure à celle d’Arabidopsis
thaliana (un polymorphisme toutes les 30 paires de
bases) et celle du blé sauvage (Triticum dicoccoides),
ces trois dernières espèces étant autogames.
Allogamie et diversité génétique
E
n général, les espèces allogames présentent une plus
grande diversité génétique que les espèces autogames,
résultat prédit par la théorie neutre de l’évolution sur
la base du fait qu’un individu allogame (et diploïde)
peut stocker à chaque locus deux allèles de deux parents
différents, tandis qu’un individu issu d’une auto-
fécondation hérite le plus souvent de deux copies du
même allèle, du même parent. En d’autres termes, c’est
la taille du réservoir utilisé pour stocker l’information
allélique qui apparaît déterminante pour expliquer
le niveau de diversité d’une espèce. On parle de taille
génétique efficace.
L
es polymorphismes neutres sont donc de bons outils
pour comprendre comment la diversité s’accumule dans
une espèce ou dans ses populations et pour faire des
prédictions sur le niveau global de diversité que l’on
peut s’attendre à trouver à un locus quelconque. Les
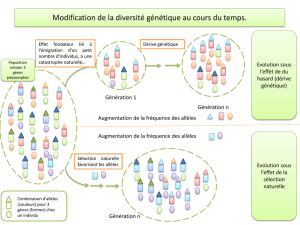
variants neutres issus des mutations (allèles) appa-
raissent constamment dans les populations. Leur fré-
quence évolue selon des processus aléatoires au sein
des populations sous l’effet de l’échantillonnage de
gamètes pour passer d’une génération à l’autre (dérive
génétique). Ils peuvent ainsi se fixer ou bien disparaître ;
en raison de leur très faible fréquence, la plupart des
mutants apparus à une génération n’ont que peu de
chances d’être retrouvés dans la génération suivante.
Plus le taux de mutation est élevé à un locus, plus le
nombre d’allèles à ce locus va être important.
Le rôle de la démographie
L
a démographie est l’autre processus important qui
module le niveau de diversité. Plus l’espèce compte
d’individus, plus sa taille génétique efficace est grande
et plus elle peut stocker d’allèles sans risquer de les
perdre par la dérive génétique. Ainsi, les espèces dis-
tribuées sur une large aire depuis une longue période
ont en général un niveau de diversité plus élevé que des
espèces peu répandues.
Encadré 1 - Les marqueurs moléculaires
Pour révéler les variations, appelées polymorphismes, au
niveau du génome, différents types de marqueurs molécu-
laires sont utilisés. Les premiers ont été les isozymes : cer-
taines enzymes existent sous différentes formes alléliques
distinguables par électrophorèse. Les marqueurs ADN ont
pris le relais et de nombreuses techniques de révélation ont
vu le jour. Les polymorphismes révélés sont de plus en plus
nombreux dans le génome, de plus en plus faciles à utiliser.
Aujourd'hui on utilise principalement les microsatellites, les
SNP, les AFLP et les DArT.
•Les microsatellites sont de courtes séquences (en géné-
ral deux à quatre bases) répétées en tandem dans de nom-
breux sites au sein d'un génome. Leur spécificité d'empla-
cement est utilisée grâce à des amorces spécifiques. Leur
polymorphisme repose sur le nombre de répétitions d’un
motif répété. Selon les espèces, les microsatellites sont
suffisamment nombreux pour permettre une bonne couver-
ture du génome. Leur taux de mutation élevé les rend très
polymorphes. Ils sont utilisés pour analyser la structure
fine et la dynamique des populations (étude de parenté,
évolution récente).
•Les AFLP (
amplified fragment length polymorphisms
),
révélés par électrophorèse, ou les DArT (
diversity array tech-
nology
), révélés par hybridation sur puce à ADN, sont issus
de la comparaison de représentations réduites du génome
des individus analysés. Il s'agit de marqueurs anonymes
(non ciblés) et généralement dominants, se prêtant moins
facilement à la génétique des populations, mais ils ont
l’avantage d’être nombreux, peu onéreux et de pouvoir s'ap-
pliquer à toute espèce sans investissement de séquençage
préalable. Simples et rapides d'application, leur bonne cou-
verture du génome les rend propices aux travaux de carto-
graphie génétique.
•Les SNP (
single nucleotide polymorphisms
) sont la forme
la plus simple et la plus fréquente de polymorphisme. Ils
correspondent à des variations de nucléotides au sein d’une
séquence connue et localisée sur le génome, en général un
gène d’intérêt. Pour l’instant, ils sont restreints à quelques
espèces bénéficiant d’importants efforts de génomique :
maïs,
Arabidopsis
, riz, blé, notamment. Chers à révéler, ils
devraient cependant bénéficier rapidement de nouvelles
technologies. Pour ces espèces, on disposera de SNP par
dizaines de milliers pour des analyses nécessitant une cou-
verture du génome très complète. Une application impor-
tante sera de déterminer la diversité allélique des gènes que
l’on pense impliqués dans des caractères adaptatifs, et de
repérer les formes les plus favorables.
*1Dans une plante allogame,
l'ovule d’une plante est
fécondé par du pollen produit
par une autre plante,
par opposition à « autogame »
(le pollen provient de la même
plante que l’ovule).
05-1 - David 13/04/06 9:31 Page 23

BIOFUTUR 266 • MAI 200624
Dossier
Génome et diversité ches les plantes cultivées
T
outefois, si l’espèce a connu une forte réduction de sa
démographie dans un passé récent, sa diversité est
réduite, et on parle alors de goulot d’étranglement. Ce
phénomène de goulot est fréquent lors du passage des
formes sauvages aux formes cultivées au cours de la
domestication. Le goulot est également drastique
lorsque l’espèce est issue d’un événement rare de poly-
ploïdisation. Sa mise en évidence avec des marqueurs
moléculaires a relancé l’intérêt de l’utilisation des popu-
lations sauvages, de nombreux allèles à des loci d’in-
térêt pouvant être disponibles dans les formes sauvages.
À
l’inverse, lorsque la population est en expansion, les
allèles rares sont moins fréquemment perdus lors du
passage d’une génération à l’autre, et la diversité aug-
mente. Ainsi, l’observation d’un excès d’allèles rares
fait penser qu’Arabidopsis thaliana est en expansion
démographique et qu’elle a reconquis récemment
l’Europe après la dernière glaciation à partir d’un foyer
se situant en Asie.
Les apports de l’informatique
L
es progrès des méthodes statistiques et l’accrois-
sement vertigineux de la puissance de calcul des ordi-
nateurs ont permis l’analyse massive de nouvelles
données moléculaires. Par exemple, regrouper les indi-
vidus partageant les mêmes combinaisons d’allèles à
de nombreux loci a permis de retracer l’histoire et
les voies de dispersion des plantes. Pour reprendre
l’exemple d’A. thaliana, un gradient de dispersion
depuis l’Asie jusqu’à la Méditerranée apparaît pro-
bable, ainsi que l'existence d’un refuge glaciaire en
Espagne-Maroc. Ce refuge méditerranéen, comme celui
se situant dans les Balkans, a également été clairement
démontré pour de nombreuses autres espèces de plantes
(comme le chêne par exemple) et de mammifères grâce
à l’étude de la diversité moléculaire.
Des outils pour rationaliser
l’exploration de la diversité
À
partir de ces informations sur la structuration de
la diversité, il est désormais possible d’améliorer les
stratégies d’exploration de la diversité génétique pour
des caractères difficiles ou coûteux à mesurer. L’idéal
est de disposer du plus petit échantillon possible rece-
lant la plus grande diversité génétique possible en
moyenne sur l’ensemble du génome. Un tel échantillon
reçoit le nom de core collection en anglais, de collec-
tion essentielle en français.
D
es simulations ont montré que des marqueurs neutres
pouvaient être efficaces pour construire ces échantillons
à condition que les populations échantillonnées de l’es-
pèce soient relativement structurées génétiquement
et isolées les unes des autres. Dans ce cas, l’informa-
tion de diversité à un locus peut se révéler efficace pour
prédire la diversité à d’autres loci même non physi-
quement proches.
A
ppliquée à Arabidopsis thaliana, cette stratégie a per-
mis de définir un lot de 48 lignées homozygotes fixées
(accessions) qui est maintenant utilisé comme échan-
tillon de référence pour étudier la diversité naturelle
de l’espèce pour des caractères tels que la résistance au
froid ou à la sécheresse
(figure 1)
. L’objectif est de détec-
ter les gènes et leurs allèles impliqués dans de nombreux
caractères et ainsi de mieux comprendre comment une
espèce comme A. thaliana s’adapte aux conditions très
variables trouvées sur son aire de répartition, des contre-
forts de l’Himalaya aux îles du Cap vert.
C
es lignées sont maintenues et régénérées avec le plus
grand soin par l’Inra de Versailles, et distribuées à la
communauté internationale. Une base de données et
un site web permettent de centraliser et rendre acces-
sible les informations acquises
(1)
.
Figure 1 Origine géographique de la
core collection
d’
Arabidopsis
Distribution mondiale des accessions d'
Arabidopsis thaliana
disponibles dans les centres de ressources (points rouges).
Les 24 accessions identifiées en jaune correspondent à une
core collection
représentant l'essentiel de la diversité connue
de l'espèce.
© D.R.
(1) http://dbsgap.versailles.
inra.fr/vnat
05-1 - David 13/04/06 9:31 Page 24

BIOFUTUR 266 • MAI 2006 25
D
’énormes efforts de description de la diversité naturelle
basés sur l’analyse des séquences de gènes ont été entre-
pris sur quelques espèces, la plupart d’importance agro-
nomique, financés surtout par les États-Unis à travers
les soutiens de la NSF (National Science Foundation).
Sur Arabidopsis, 876 fragments de gènes ont été séquen-
cés sur un échantillon de 96 individus. Sur le maïs, un
effort similaire a été rendu public en 2005 et d’impor-
tants résultats sont attendus sur le riz et le blé.
L
e blé et la vigne ont également reçu une attention parti-
culière de l’Inra, pour décrire leur diversité sur un
nombre de marqueurs microsatellites important. Ainsi,
l’ensemble des cépages conservés au Centre de ressources
biologique de Montpellier a été génotypé, de même que
5 000 lignées de blés conservées au CRB de Clermont-
Ferrand représentatives de la diversité mondiale.
Programmes et consortiums
A
u-delà de ces efforts de recherche par séquençage
haut-débit sur les gènes eux-mêmes, la communauté
internationale a mis en place le challenge program
« Generation »
(2)
. C’est un programme du Groupe
consultatif pour la recherche agronomique interna-
tionale qui focalise le développement et l'application
de la génomique vers l'exploitation de la diversité géné-
tique pour l’amélioration des plantes, en particulier
celle présente dans les grandes collections internatio-
nales de plantes cultivées et leurs parents sauvages.
L
es organismes de recherche agronomique français sont
membres du consortium fondateur sous la bannière
d'Agropolis à Montpellier. Les principales cultures
vivrières sont concernées (maïs, blés, riz, orge, sorgho,
mil et millets divers, haricots, divers pois, arachide,
niébé, manioc, pomme de terre, igname, bananier, coco-
tier) et le caractère cible prioritaire est la tolérance à la
sécheresse. Il s'agit d'analyser en détail la diversité géné-
tique, de déployer des analyses de génomique compa-
rative, de gérer et exploiter les données produites dans
un cadre bioinformatique performant et d'intégrer les
résultats de recherche dans le développement de nou-
velles variétés, le tout en partenariat étroit avec les uti-
lisateurs dans les pays du Sud.
Repérer la variation qui cause
les différences phénotypiques, un défi
É
merge ainsi une des questions les plus excitantes de la
génétique en ce début du XXIesiècle. Dans l’ensemble de
la diversité observée au niveau de l’ADN, quelle part
de la variation tient un rôle adaptatif ? Peut-on identifier
les bases nucléotidiques qui sont directement responsables
de la variation phénotypique ? L’analyse de la diversité
naturelle peut-elle nous aider à comprendre comment se
construit un phénotype à partir de variations moléculaires
à différents loci ?
C
ette question mobilise aujourd’hui une grande partie de
la communauté des généticiens, quel que soit l’organisme
sur lequel ces derniers travaillent, de l’humain jusqu’aux
plantes. Cela consiste à chercher l’aiguille du polymorphisme
causal dans la botte de foin du polymorphisme neutre.
L’approche QTL
U
ne des approches expérimentales employées pour ces
études consiste à rechercher les QTL
*2
. Les cartes géné-
tiques permettent de suivre la ségrégation des gènes dans
les descendances en couvrant l'ensemble du génome.
Tout comme le polymorphisme moléculaire des gènes,
ou un caractère phénotypique simple (la couleur, par
exemple), les caractères quantitatifs (taille de la plante,
nombre de graines, etc.) peuvent être mesurés dans
la descendance d’un croisement entre deux plantes. On
peut ainsi déterminer si le ou les caractères quantitatifs
étudiés sont associés de manière statistiquement signi-
ficative à une zone chromosomique particulière repé-
rée par les marqueurs moléculaires qui s’y trouvent, et
dont la ségrégation a aussi été suivie dans cette même
descendance.
C
ela donne un pouvoir de résolution supérieur à celui
de l'analyse des ségrégations génétiques dans les des-
cendances, qui permet de repérer les différentes zones
du génome impliquées dans l'élaboration de caractères
quantitatifs complexes et de les classer selon leur effet
sur le caractère.
Encadré 2 - Des polymorphismes « candidats »
dans le gène
opaque-2
chez le sorgho
Opaque-2
est un gène identifié
d'abord chez le maïs, à partir d'une
mutation qui produit un grain opaque.
Il agit par activation de la transcrip-
tion de certains gènes et régule en
particulier l'expression d'un gène qui
produit une zéine (une classe de pro-
téines de réserve du maïs,
Zea mays
)
de 22 kDa. Il fait partie d'une liste de
gènes candidats pris en compte dans
un projet associant GABI et Géno-
plante, les programmes nationaux de
génomique végétale allemands et
français, pour expliquer les variations
de la teneur en protéines du grain
chez les céréales.
Chez le sorgho, les travaux sont
conduits par Lucio Alencar, cher-
cheur de l'université catholique de Brasilia, et Monique Deu du Cirad.
Opaque-2
a été amplifié
(sur une base de huit fragments, a à h) et séquencé pour un ensemble de variétés représen-
tatives de la diversité du sorgho, révélant au total plus de 50 SNP et plusieurs indels (inser-
tions/délétions) le long des 4,3 kb analysés, qui recouvrent une région amont de 1,5 kb et la
partie codante de 2,8 kb, faite de six exons (régions codantes du gène) et cinq introns (non
codants).
En utilisant 188 variétés pour lesquelles on dispose de plus de 80 % de la séquence (les don-
nées sont incomplètes pour le segment f), on relève un déséquilibre de liaison fort, avec seu-
lement 22 haplotypes différents (sur un total de 256 possibles) qui se répartissent en six
groupes d'effectif important, appartenant à deux grands types très différents (groupes 1 à 5
et groupe 6). Certains haplotypes sont spécifiques d'une race (comme les
Guinea margariti-
ferum
) ou d'une provenance (comme l'Afrique australe) et donc révèlent essentiellement des
éléments de structure. D'autres se répartissent sur un grand ensemble de types variétaux et
l'effet de leur présence/absence sur le phénotype des variétés peut être testé efficacement.
Les mêmes variétés sont évaluées pour de nombreuses caractéristiques du grain.
© LUCIO ALENCAR
*2
Quantitative trait loci
, zone
chromosomique qui contrôle
la valeur d'un caractère.
(2) CPG, www.generationcp.org
© D.R.
05-1 - David 13/04/06 9:31 Page 25

BIOFUTUR 266 • MAI 200626
Dossier
Génome et diversité ches les plantes cultivées
I
l existe aujourd'hui d'importants répertoires de QTL
sur de nombreuses espèces. Les réussites les plus spec-
taculaires sont probablement le repérage d'allèles favo-
rables (quantitative trait alleles) dans les espèces
sauvages pour des caractères pourtant supérieurs chez
l'espèce cultivée, ainsi que l'identification de gènes nou-
veaux grâce au clonage de QTL. Après une quinzaine
d'années de travaux sur l'exploitation des espèces sau-
vages pour l'amélioration de la tomate, Steve Tanksley
de l'université de Cornell aux États-Unis constate qu'un
cinquième des loci identifiés à partir d'une analyse QTL
présente une forme allélique plus favorable dans le
parent sauvage que dans le parent cultivé et que la
source est loin de se tarir, chaque nouvelle espèce sau-
vage utilisée, même éloignée, apportant son lot de nou-
veaux allèles
(3)
.
S
ur le riz, la disponibilité de la séquence du génome
facilite le clonage de QTL. L'équipe de Masahiro Yano
au National Institute of Agrobiological Sciences de
Tsukuba au Japon est parvenue à cloner plusieurs QTL
dans le cadre d'une analyse systématique de la diversité
naturelle de contrôle de la floraison
(4)
. Elle a ainsi révélé
des gènes orthologues de gènes connus chez Arabidopsis
thaliana (Hd1, RFT1), mais aussi des gènes sans équi-
valent connu aujourd’hui (Ehd1, Hd5, Hd6, Lhd4).
La génétique d’association
S
e développent également différentes approches regrou-
pées sous le terme de génétique d'association. La pre-
mière consiste à cibler l'analyse de la diversité
moléculaire au niveau de gènes candidats, de façon à
inventorier toutes les formes alléliques présentes dans
un échantillon donné
(encadré 2)
, et à rechercher les
associations apparentes entre profil moléculaire et
expression phénotypique. On espère ainsi valider le
fait qu'un gène candidat donné est important sur le
plan fonctionnel et repérer les formes alléliques les plus
« favorables »
(encadré 3)
.
L
'approche complémentaire procède au contraire à un
balisage complet et sans a priori du génome, et tire
avantage de ce qu'on appelle le déséquilibre de liaison
(DL). Celui-ci vient du fait que les gènes sont liés entre
eux par l’organisation physique des chromosomes. Se
créent alors des propriétés d’association particulières
entre les polymorphismes à des loci différents, avec
une dépendance statistique globalement plus forte entre
loci liés sur le même chromosome
(figure 2)
.
P
ar exemple, entre deux loci présentant chacun un
polymorphisme à deux allèles (A/a et B/b), l'allèle A
au premier locus sera plus fréquemment associé à l’al-
lèle Bqu’à l’allèle b au second locus (avec l'associa-
tion complémentaire entre l'allèle aet l'allèle b). Si les
allèles Bet bsont in fine responsables d’une part de la
variation d’un caractère, par exemple si les individus
BB sont plus précoces que les individus bb, alors les
individus AA seront aussi plus précoces que les aa
puisque Aest associé statistiquement à B.
L
a probabilité d’avoir un marqueur (locus A) lié au
polymorphisme causal (locus B) augmente avec la den-
sité de marquage et l’étendue du DL autour du locus B.
Le déséquilibre de liaison se crée par la mutation, la
dérive génétique, la sélection et la différenciation entre
populations, et il est cassé par la recombinaison lors
de la méiose chez les individus hétérozygotes. Il en
résulte une possibilité de cartographier le génome en
utilisant des échantillons de plantes dont on connaît
l’histoire évolutive.
L
es recombinaisons s’accumulent au cours des généra-
tions et l’étendue du DL finit par décroître pour arri-
ver à son point d’équilibre. L’étendue cartographique du
DL varie considérablement d’une espèce à l’autre. Le DL
s'étend couramment au-delà de la centaine de kilobases
(kb) chez les variétés traditionnelles de céréales, de 50 à
250 kb chez A. thaliana, et quelques kilobases chez le
maïs. Il peut parfois atteindre plusieurs dizaines de centi-
morgans
*3
(un quart de chromosome) chez les cultivars
modernes issus d'amélioration génétique récente.
Quelques résultats
A
insi, les premiers résultats sur le maïs ou le riz sont
encourageants sur des régions ciblées. Chez le maïs,
un polymorphisme a été trouvé au sein du gène
Dwarf8 comme potentiellement responsable de la
durée de développement. De même, Susan McCouch
(université Cornell) conclue de ses études sur la
Encadré 3 - La diversité génétique du riz
Le riz, culture vivrière de premier rang mais aussi modèle pour l'analyse génétique des céréales,
est la deuxième plante à avoir eu son génome séquencé. Le Génoscope a assuré le séquen-
çage de l’un de ses 12 chromosomes. L'espèce asiatique assure plus de 95 % de la pro-
duction. Toutes les générations de marqueurs moléculaires s'accordent pour lui attribuer une
origine multiple, avec des domestications en Chine (type
japonica
) comme en Inde (type
indica
), qui expliquerait son extraordinaire diversité adaptative et agromorphologique.
Grâce à la disponibilité des séquences de plusieurs variétés, on dispose de centaines de mil-
liers de SNP couvrant l'ensemble du génome. Les estimations de DL montrent des asso-
ciations s'étendant jusqu'à 100 kb, sans compter le DL d'introgression qui caractérise les
interfaces issues de recombinaison entre groupes variétaux.
La variation de séquence au niveau de gènes impliqués dans des caractères importants comme
la qualité du grain (gène
waxy
, dont un allèle rend le riz glutineux) permet de jumeler ana-
lyses biochimiques, génétiques, évolutives et ethnologiques. Ainsi, la mutation respon-
sable du grain glutineux induit l'absence d'amylose (une composante particulière de l'amidon)
par défaut d'épissage de l'ARN messager correspondant. Elle est apparue et a été sélec-
tionnée dans le type
japonica
avant de se répandre dans le type
indica
sous l'action des
flux polliniques et de la sélection opérée par les riziculteurs d'Asie du Sud-Est.
De la même façon, l'arôme de certains riz est essentiellement contrôlé par une délétion de
huit paires de bases dans un gène de bétaïne aldéhyde déshydrogénase. Il semble qu'elle ne
soit apparue qu'une fois et se soit répandue au sein de différents types variétaux pour don-
ner les différents riz basmati de l'Inde et du Pakistan et les riz parfumés de Thaïlande (
indica
)
ou des Philippines (
japonica
).
Cigalon – Camargue
Basmati 370 – Inde, Pakistan (parfumé)
Inca (Ciat-Cirad)
Ram tulasi, Inde
Khao Youak, Laos (
waxy
)
Irat 216 – Cirad
Khao Dawk Mali 105
Thaïlande, jasmin (parfumé)
© J.-C. G.
*3Distance génétique,
1 cM équivaut à 1 %
de recombinaison pendant
la méiose.
(3) Frany A
et al.
(2003)
Genome
46,235-43
(4) Li Z
et al.
(2003)
Plant Phys Biochem
42, 1-6
05-1 - David 13/04/06 9:31 Page 26
 6
6
1
/
6
100%