Lire l`article complet

Diabète
Dossier
268
Métabolismes Hormones Diabètes et Nutrition (VII), n° 6, novembre/décembre 2003
Les diabètes monogéniques : caractéristiques
cliniques et génétiques du diabète de type MODY
et du diabète mitochondrial
S. Boullu-Sanchis*
L’
hétérogénéité clinique du dia-
bète est l’un des éléments clés
qui ont fait suspecter une ori-
gine génétique de la maladie. Le
regroupement pour une analyse géné-
tique de patients qui présentaient les
mêmes caractéristiques cliniques a
permis d’aboutir à la mise en évi-
dence de gènes responsables d’un
diabète monogénique tel que le dia-
bète de type Maturity-Onset Diabetes
of the Young (MODY).
Les dix dernières années ont été riches
en données nouvelles concernant la
génétique des diabètes non insulino-
dépendants, entraînant une modifi-
cation de la classification des dia-
bètes en 1997 sur proposition de
l’American Diabetes Association. En
effet, celle-ci comporte maintenant
une classe spécifique de diabètes
secondaires à des anomalies géné-
tiques de l’insulinosécrétion (diabètes
de type MODY et diabète mitochon-
drial) ou à des anomalies génétiques
de l’insulinosensibilité (syndromes
rares d’insulinorésistance extrême,
diabète lipoatrophique) et comporte
une rubrique “Entités autres” qui
laisse la place à de nouvelles données.
Les deux types de diabète monogé-
nique les plus fréquents sont les dia-
bètes de type MODY à transmission
autosomique dominante et le diabète
mitochondrial à transmission mater-
nelle associé à une surdité, appelé
aussi Maternally Inherited Diabetes
and Deafness (MIDD). Les diabètes
monogéniques représentent 2 à 5 %
des diabètes non insulinodépendants.
Les diabètes de type
MODY : généralités
Les diabètes de type MODY sont
caractérisés, selon la définition ini-
tiale, par un début précoce de la
maladie, avec un âge au moment
du diagnostic inférieur à 25 ans, un
caractère non insulinodépendant
durant au moins les deux premières
années de la maladie et une trans-
mission de type autosomique domi-
nant. En raison des anomalies géné-
tiques connues et de l’étude des
caractères phénotypiques des patients
atteints, la présence d’une anomalie
primaire de l’insulinosécrétion a été
ajoutée à la définition (1).
Actuellement, des mutations de six
gènes différents sont identifiées. Il
s’agit du gène codant la glucokinase
(GCK) et de cinq gènes codant des
* Attachée aux hôpitaux de Strasbourg, service
d’endocrinologie, CHU de Strasbourg.
▲
▲Les diabètes monogéniques, dont les deux principaux types sont le
diabète de type MODY et le diabète mitochondrial, représentent 2 à 5 %
des diabètes non insulinodépendants.
▲
▲Six gènes sont actuellement connus comme étant à l’origine du diabète
de type MODY. Il s’agit de mutations du gène de la glucokinase (enzyme
clé du métabolisme glucidique des cellules pancréatiques et hépato-
cytaires) et de cinq gènes codant pour des facteurs de transcription
nucléaire participant au développement du pancréas endocrine (HNF-1a,
HNF-4a, HNF-1b, IPF-1 et Neuro-D1).
▲
▲Il existe une hétérogénéité clinique parmi les différents diabètes de
type MODY, notamment entre le MODY 2 (diabète peu sévère en termes
de complications) et le MODY 3.
▲
▲La réalisation du test génétique est à envisager chez les patients pré-
sentant un phénotype évocateur : diabète non insulinodépendant surve-
nant avant 30 ans, dans un contexte familial et en l’absence de surpoids.
Les sujets “à risque” sont également les enfants hyperglycémiques non
insulinodépendants ou qui présentent une “lune de miel” prolongée
(MODY 2) ; les femmes minces qui présentent un diabète gestationnel
(MODY 2 ou 3) ; les patients souffrant de malformations génito-rénales
et d’un diabète non insulinodépendant familial (MODY 5).
▲
▲Le diabète mitochondrial est lié à une mutation de l’ARN. La transmis-
sion de la mutation se fait selon un mode matrilinéaire.
▲
▲Le diabète mitochondrial se caractérise par l’association d’un diabète,
d’une surdité familiale et d’une dystrophie maculaire réticulée.
points FORTS

Métabolismes Hormones Diabètes et Nutrition (VII), n° 6, novembre/décembre 2003
Diabète
Dossier
269
facteurs de transcription nucléaire
participant au développement du pan-
créas endocrine : Hepatocyte Nuclear
Factor-1
a
(HNF-1a), Hepatocyte
Nuclear Factor-4
a
(HNF-4a), Hepa-
tocyte Nuclear Factor-1
b
(HNF-1b),
Insulin Promotor Factor-1 (IPF1) et
Neurogenic Differentiation Factor-1
(Neuro-D1). Le tableau I résume les
principales caractéristiques des six
sous-types actuellement connus de
diabète de type MODY.
Les caractéristiques
phénotypiques
des diabètes
de type MODY 2 et 3
La prévalence des différents sous-
type de MODY varie selon les popu-
lations : en effet, le MODY 2 repré-
sente environ 60 % des cas de MODY
en France alors qu’il en représente
seulement 20 % en Angleterre. Toute-
fois, malgré ces différences, les deux
formes les plus fréquentes sont le
MODY 2 et le MODY 3. Les quatre
autres sous-types sont rares et
concernent le plus souvent quelques
familles à travers le monde. Nous
nous attacherons donc à décrire plus
précisément les deux formes les
plus fréquentes de diabète de type
MODY, dont les principales carac-
téristiques sont reprises dans le
tableau II.
Les caractéristiques
phénotypiques du MODY 2
La mise en évidence d’une relation
entre une mutation hétérozygote du
gène codant la glucokinase (GCK)
et un diabète de type MODY remonte
à 1993 et correspond au premier sous-
type génétique de MODY décrit (2).
La glucokinase est une enzyme clé du
métabolisme intracellulaire du glu-
cose des cellules insulinosécrétrices
et hépatocytaires. Cette enzyme, qui
catalyse la phosphorylation du glu-
cose en glucose 6 phosphate, agit
comme un glucose sensor pour les
cellules bpancréatiques. Les muta-
tions hétérozygotes du gène codant
pour la GCK entraînent une dimi-
nution de la phosphorylation du glu-
cose. Cette modification induit une
augmentation du seuil glycémique
stimulant la libération d’insuline et
entraîne une diminution de la quan-
tité d’insuline libérée. Actuellement,
plus de 130 mutations du gène GCK
à l’origine d’un tableau phénotypique
de MODY 2 ont été décrites dans
différentes populations (3).
L’hyperglycémie à jeun est modérée,
le plus souvent entre 5,5 et 9 mmol/l,
MODY 1 MODY 2 MODY 3 MODY 4 MODY 5 MODY 6 MODY X
Génotype HNF 4aGCK HNF 1aIPF-1 HNF 1bNeuroD1 ?
Localisation 20q 7p 12q 13q 17q – ?
chromosomique
Début de adolescence In utero adolescence adolescence adolescence adolescence ?
hyperglycémie
Sévérité de Progressive, modéree, Progressive, Non connu Progressive, Progressive, variable
l’hyperglycémie peut être détérioration peut être peut être peut être
sévère mineure sévère sévère sévère
avec âge
Sévérité +++ + +++ ++ +++ +++ variable
des complications
microvasculaires
Fréquence < 1 % 60 % 20 % < 1 % < 1 % < 1 % 15 à 20 %
en France
Abréviations : HNF :
Hepatic Nuclear Factor,
GCK :
glucokinase,
IPF-1 :
Insulin Promoter Factor-1,
Neuro-D1 :
Neurogenic Differentiation Factor-1.
Tableau I. Caractéristiques des 6 sous type de diabète de type MODY.
Tableau II. Principales caractéristiques cliniques des MODY 2 et 3.
Caractéristiques des 2 principaux MODY
•Différents gènes – différents diabètes type MODY
– début in utero
– hyperglycémie à jeun modérée
– traitement : ADO ou diététique
– complications peu sévères
– début adolescence/jeune adulte
– aggravation progressive
de l’hyperglycémie
– traitement : 1/3 ADO, 1/3 diététique,
1/3 insuline
– complications fréquentes et sévères
MODY 2 GCK MODY 3 HNF-1 ?
MODY

Métabolismes Hormones Diabètes et Nutrition (VII), n° 6, novembre/décembre 2003
Dossier
Diabète
270
et cette anomalie débute in utero.
En effet, il a été mis en évidence
que les nouveau-nés qui présentaient
des mutations du gène codant pour
la glucokinase avaient un poids de
naissance inférieur d’environ 500 g
à celui des nouveau-nés non atteints.
Ce “petit poids” était alors directe-
ment en relation avec une insulino-
pénie déjà présente in utero.
On observe une altération modérée
de l’insulinosécrétion, associée à
une augmentation souvent modérée
de la glycémie à la 2eheure après
charge orale en glucose. Cette alté-
ration de l’insulinosécrétion est
relativement stable dans le temps ;
en effet, l’équilibre glycémique est
souvent, et pendant longtemps, bien
contrôlé par les anti-diabétiques
oraux, voir par les mesures hygiéno-
diététiques seules. Les patients insu-
linorequérants sont relativement
rares et représentent environ 2 %
des patients MODY 2.
Les complications micro- ou macro-
vasculaires sont relativement rares
dans le MODY 2. Ce pronostic rela-
tivement favorable du diabète de type
MODY 2 est un des arguments en
faveur de la réalisation du test géné-
tique. En effet, le diagnostic molé-
culaire permet alors de préciser le
pronostic de la maladie, mais égale-
ment de guider le dépistage familial.
Dans le MODY 2, la pénétrance du
gène est complète, c’est-à-dire que
tous les sujets porteurs de la muta-
tion sont hyperglycémiques. Le
pronostic relativement favorable
de ce type de diabète tempère cette
situation.
La présence de mutations homo-
zygotes du gène codant pour GCK
a été associée au diabète néonatal
sévère (4). En effet, de telles muta-
tions ont été mises en évidence chez
des nourrissons qui avaient présenté
un “diabète néonatal” : les mutations
retrouvées n’avaient encore jamais
été décrites. Ces auteurs proposent
l’hypothèse selon laquelle certains
diabètes néonataux transitoires cor-
respondraient à un diabète de type
MODY 2 qui se révélerait à l’âge
adulte.
Les caractéristiques
phénotypiques du MODY 3
Le MODY 3 est lié à une mutation
hétérozygote du gène codant un fac-
teur de transcription nucléaire, le
HNF-1a. Le HNF-1aest exprimé
dans de nombreux tissus et ne repré-
sentait pas a priori un gène candidat
du diabète. Une relation entre une
mutation du gène codant HNF-1a
et un diabète de type MODY a été
mise en évidence suite à la décou-
verte d’une liaison au niveau du
chromosome 12q au cours d’études
de criblage du génome chez des
patients présentant un phénotype de
MODY mais n’ayant pas de muta-
tion du gène GCK (5). Actuellement,
une centaine de mutations diffé-
rentes du gène codant HNF-1aont
été décrites dans différentes popula-
tions. Le MODY 3 représente la pre-
mière cause de MODY (plus de 60 %
des cas) en Angleterre et la deuxième
en France (environ 20 %).
La pénétrance des mutations du gène
HNF-1aest incomplète (76 %) à
l’âge de 30-40 ans, au contraire du
MODY 2, ce qui explique les variabi-
lités phénotypiques observées d’une
famille à l’autre. Si certains patients
qui sont porteurs de la mutation ne
présentent pas d’hyperglycémie à
jeun, on observe en revanche toujours
une altération de la réponse insulino-
sécrétoire à une charge en glucose.
Le début des anomalies métaboliques
est post-pubertaire et l’âge lors du
diagnostic est le plus souvent infé-
rieur à 25 ans. Toutefois, ce type de
diabète a été mis en évidence après
l’âge de 30 ans chez 26 % des patients
finlandais de la Botnia Study.
À l’inverse de ce qui est observé dans
le MODY 2, les anomalies de la
sécrétion d’insuline chez les patients
MODY 3 s’installent progressive-
ment, et s’aggravent aussi progres-
sivement avec les années, en se rap-
prochant de ce qui est constaté dans
le diabète de type 1 lent. Les com-
plications de la micro-angiopathie
(rétinopathie et néphropathie) sont
plus fréquentes et sévères chez les
patients MODY 3, par comparaison à
la relative “bénignité” du MODY 2,
probablement du fait de l’altération
progressive et sévère de l’insulino-
sécrétion.
Une particularité phénotypique des
patients porteurs d’un diabète de type
MODY 3 est représentée par une
sensibilité aux sulfamides hypogly-
cémiants. En effet, il a été rapporté
quelques cas d’hypersensibilité à ces
médicaments, avec survenue d’un
déséquilibre glycémique très sévère
lorsque ces traitements étaient stop-
pés. Ces éléments semblent justifier
la prescription en première intention
de ce type d’antidiabétiques oraux
et invitent par ailleurs à la vigilance
vis-à-vis du risque d’hypoglycémie
lors de l’introduction du traitement.
Les autres formes
de diabète MODY
Le MODY 1
Le diabète de type MODY 1 est une
forme rare de MODY liée à une muta-
tion du gène codant pour HNF-4a.
Ce facteur de transcription régule
HNF-1a, et la présentation phéno-
typique du MODY 1 est relativement
proche de celle du MODY 3. Les
mutations ont été décrites dans une
“super” famille américaine de dia-
bète de type MODY.
Le MODY 4
L’IPF1 est un facteur majeur néces-
saire au développement du pancréas
endocrine et à la régulation de la trans-
cription des gènes impliqués dans le
fonctionnement des cellules bpan-
créatiques. La première description
d’une mutation du gène codant ce
facteur de transcription concernait
un nouveau-né présentant une hyper-
glycémie majeure liée à une agénésie
du pancréas (6). Cet enfant présentait
une mutation homozygote du gène
de l’IPF1, qui est situé au niveau du
chromosome 13q. L’analyse clinique
familiale a révélé un diabète gesta-
tionnel chez sa mère et un diabète
non insulinodépendant chez son père.
Les parents étaient porteurs hétéro-
zygotes de la mutation. Cette mutation

Métabolismes Hormones Diabètes et Nutrition (VII), n° 6, novembre/décembre 2003
Diabète
Dossier
271
du gène codant IPF1 a été retenue
comme celle liée au MODY 4, forme
rare de MODY dont la sévérité est
intermédiaire entre celle du MODY 2
et celle du MODY 3 en termes de
complications micro-vasculaires.
Le MODY 5
Le MODY 5 correspond à une asso-
ciation entre un phénotype clinique
de diabète de type MODY et une
mutation du gène codant HNF-1b.
Ce facteur de transcription régule
l’expression, comme HNF-4a,de
HNF-1a. Le MODY 5 représente une
cause rare de diabète de type MODY,
dont la particularité réside dans l’asso-
ciation de malformations rénales et/ou
génitales à caractère familial à un
diabète de type MODY.
Chez le rat, HNF-1bjoue un rôle
dans le développement du rein.
Chez l’homme, une mutation du
gène codant HNF-1ba été mise en
évidence : le phénotype associait un
diabète et des kystes rénaux mul-
tiples de transmission familiale (7).
Plus récemment, une équipe fran-
çaise a mis en évidence de nouvelles
mutations de HNF-1bdans un
contexte clinique de kystes rénaux
et hépatiques à transmission fami-
liale et d’un diabète de type MODY.
Le MODY 6
Dernière mutation connue et décrite à
l’origine d’un diabète de type MODY,
Neuro-D1 est également un facteur
de transcription nucléaire proche de
HNF. Les caractéristiques phénoty-
piques de ce type de MODY sont
pour l’instant mal connues. La muta-
tion a été décrite dans une famille
islandaise (8) et n’a pas été retrouvée
dans d’autres populations à l’heure
actuelle.
Le diabète de type MODY,
un diabète monogénique
“très hétérogène”
Le diabète MODY, diabète monogé-
nique le plus fréquent, est en fait une
entité très hétérogène. On retrouve
une hétérogénéité génétique, avec
actuellement six gènes connus, et
probablement d’autres à venir. En
effet, on observe des familles qui
ont un phénotype clinique corres-
pondant à un diabète MODY et qui
ne présentent aucune des mutations
connues. Cela est particulièrement
net chez les patients MODY japo-
nais, dont plus de 70 % ne présen-
tent aucune des six mutations. Par
ailleurs, plusieurs mutations ont été
décrites pour les différents types de
MODY connus à ce jour : elles sont
plus de 100 pour les MODY 2 et 3.
Une hétérogénéité clinique est éga-
lement observée, hétérogénéité entre
les différents sous-types de MODY,
mais également du fait d’une péné-
trance incomplète, comme pour le
MODY 3, ce qui entraîne une varia-
bilité de l’expression phénotypique
pour une même mutation.
Le diabète mitochondrial
Au début des années 1990, J.M.W.
Van den Ouweland et al. (9) ont décrit
un syndrome associant un diabète,
une surdité et une transmission matri-
linéaire, syndrome appelé Maternally
Inherited Diabetes and Deafness
(MIDD). Ce syndrome est lié à une
mutation ponctuelle en position
3 243 de l’ADN mitochondrial, avec
le remplacement d’une adénine par
une guanine au niveau de l’ARN de
transfert de la leucine. La prévalence
de ce syndrome parmi des cohortes
de diabétiques non insulinodépen-
dants varie de 0,5 à 2,8 % selon les
populations. Le tableau III reprend
les principaux syndromes associés à
un diabète mitochondrial.
La présentation clinique du diabète
associe : un déficit de l’insulinosé-
crétion, un développement précoce de
la maladie, avec un âge moyen lors
du diagnostic compris entre 25 et
35 ans, et l’absence d’obésité, avec
un IMC inférieur à 27 kg/m2. Les
caractéristiques cliniques sont main-
tenant mieux connues, permettant
d’orienter la suspicion diagnostique.
D’après les données d’une cohorte
française de patients atteints de
diabète mitochondrial, on retient
les données cliniques suivantes (10).
L’âge moyen lors du diagnostic était
de 40 ans environ et, chez près de
50 % des patients, le diabète avait été
diagnostiqué avant l’âge de 25 ans.
Aucun des patients n’était obèse, et
40 % d’entre eux avaient un IMC
inférieur à 18,5 kg/m2. Une surdité
bilatérale neurosensorielle était pré-
sente chez pratiquement tous les
patients, et l’âge lors du diagnostic
de la surdité était très variable d’un
sujet à l’autre. L’existence d’une dys-
trophie maculaire réticulée a été mise
en évidence chez 85 % des sujets
étudiés. Les complications de type
micro angiopathie étaient peu fré-
quentes, 30 % des patients présen-
taient une hypertension et 10 % une
maladie coronaire. Ces différents élé-
ments cliniques, et plus particuliè-
rement la surdité et la dystrophie
maculaire réticulée, représentent des
arguments devant faire évoquer et
rechercher un diabète mitochondrial.
En pratique, chez qui
et pourquoi demander
un diagnostic moléculaire
de diabète monogénique ?
Les diabètes monogéniques restent
des formes rares de diabète non
insulinodépendant. Toutefois, en
Tableau III. Les diabètes mitochondriaux.
•Maternally Inherited Diabetes
and Deafness (MIDD)
✓Surdité, dystrophie maculaire,
atteintes neuromusculaires
✓Mutation 3243 ARNtLeu
✓0,5 à 2,8 % des diabètes de type 2
•Sd Keams – Sayre
•Sd de Wolfram (DI DMOAD)
•Sd MELAS

Métabolismes Hormones Diabètes et Nutrition (VII), n° 6, novembre/décembre 2003
Dossier
Diabète
272
raison des connaissances actuelles,
le diagnostic moléculaire peut être
demandé quand les circonstances
cliniques sont évocatrices.
Chez qui évoquer le diagnostic
de diabète monogénique ?
Il faut envisager ce diagnostic devant
tout patient non obèse qui présente
une hyperglycémie chronique non
insulinodépendante avant l’âge de
30 ans. Le diagnostic sera d’autant
plus évocateur s’il existe un contexte
familial de diabète. Avant de demander
l’analyse génétique, il est souhaitable
d’éliminer un diabète de type 1 lent,
ou LADA, qui reste le diagnostic
différentiel principal, en réalisant un
groupage HLA et un dosage des
anticorps anti-GAD et anti-IA2. Le
contexte clinique peut orienter plus
précisément la suspicion diagnos-
tique. En effet, la présence de kystes
rénaux évoquera un MODY 5 ; une
surdité, une transmission matrilinéaire
et/ou une dystrophie maculaire feront
évoquer un diabète mitochondrial ;
un diabète non insulinodépendant
de l’enfant ou de l’adolescent fera
envisager un MODY 2.
Pourquoi demander
un diagnostic moléculaire
de diabète monogénique ?
Le diagnostic moléculaire présente
plusieurs intérêts. En premier lieu,
celui du diagnostic étiologique, qui
permet alors de mieux préciser le
pronostic de la maladie. Cela est
d’autant plus important lorsque l’on
suspecte un diabète de type MODY
et que l’on connaît les différences
cliniques entre les MODY 2 et 3.
On peut également évoquer la notion
de dépistage familial et le caractère
“préventif” que peut avoir le dia-
gnostic moléculaire. En effet, si l’on
ne peut pas prévenir le développe-
ment du diabète chez les patients
porteurs de la mutation, on peut
retarder l’apparition des complica-
tions métaboliques en appliquant
chez ces patients des mesures pré-
ventives hygiéno-diététiques.
Conclusion
Les diabètes monogéniques sont
aujourd’hui mieux connus. Une amé-
lioration de la connaissance des traits
phénotypiques, permet d’aider les
cliniciens à identifier les sujets dia-
bétiques concernés et leurs familles.
Le diagnostic moléculaire est aujour-
d’hui possible en “pratique courante”
pour les MODY 2, 3 et 5 et pour la
mutation 3243 du diabète mitochon-
drial. La recherche de l’anomalie
génétique présente des avantages,
notamment en termes de pronostic
de la maladie et de dépistage familial.
L’hétérogénéité clinique et génétique
implique que de nouvelles données
sont à attendre dans les années à
venir, sur le plan génétique mais éga-
lement sur le plan thérapeutique.
Références essentielles*
et incontournables**
1.
** Hattersley AT. Maturity onset diabetes of the
young : clinical heterogeneity explained by genetic
heterogeneity. Diabet Med 1998 ; 15 : 15-24.
2.
** Froguel P, Zouali H, Vionnet N et al. Familial
hyperglycemia due to mutations in glucokinase :
definition of a subtype of diabetes mellitus. N Engl
J Med 1993 ; 328 : 697-702.
3.
* Permutt MA, Hattersley AT. Searching for
type 2 diabetes genes in the post-genome era.
Trends Endocrinol Metab 2000 ; 11 : 383-93.
4.
Prisco F, Iafusco D, Franzese A et al. MODY 2
presenting as neonatal hyperglycemia : a need to
reshape the definition of neonatal diabetes. Dia-
betologia 2000 ; 43 : 1331-2.
5.
Vaxillaire M, Boccio V, Philippi A et al. A gene
for maturity-onset diabetes of the young (MODY)
maps to chromosome 12q. Nat Genet 1995 ; 9 :
418-23.
6.
* Stoffers DA, Zinkin NT, Stanojevic V et al.
Pancreatic agenesis attributable to a single nucleo-
tide deletion in the human IPF1 gene coding
sequence. Nat Genet 1997 ; 15 : 106-10.
7.
Bingham C, Ellard S, Allen LI et al. Abnormal
nephron development associated with a frameshift
mutation in the transcription factor hepatocyte
nuclear factor-1beta. Kidney Int 2000 ; 57 : 898-
907.
8.
Kristinsson SY, Thorolfsdottir ET, Talseth B et al.
MODY in Iceland is associated with mutations in
HNF-1alpha and a novel mutation in Neuro-D1.
Diabetologia 2001 ; 44 (11) : 2098-101.
9.
** Van den Ouweland JMW, Lemkes HHPJ,
Ruittenbeek W et al. Mutation in mitochondrial
tRNALeu(UUR) gene in a large pedigree with mater-
nally transmitted type II diabetes mellitus and
deafness. Nat Genet 1992 ; 1 : 368-71.
10.
* Guillausseau PJ, Massin P, Dubois-
Laforgue D et al. Maternally inherited diabetes
and deafness : a multicenter study. Ann Intern
Med 2001 ; 134 (9 Pt 1) : 721-8.
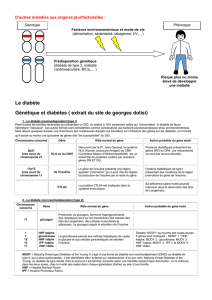
1. Six gènes sont connus actuellement comme étant à l’origine d’un diabète de type MODY. Vrai ou faux ?
2. Le diabète mitochondrial est lié à une transmission paternelle de la maladie. Vrai ou faux ?
3. Le diabète de type MODY 2 est caractérisé par un trouble sévère de l’insulinosécrétion et est associé
à des complications microvasculaires fréquentes et graves. Vrai ou faux ?
4. L’association de kystes rénaux et/ou hépatiques et d’un diabète de type MODY est évocatrice d’un MODY 5.
Vrai ou faux ?
1. Vrai. 2. Faux. 3. Faux. 4. Vrai.
Auto-test
1
/
5
100%