1 HEMATOLOGIE : Notion d`Hématologie.

1
HEMATOLOGIE : Notion d’Hématologie.
I) Origine des éléments figurés du sang :
Le sang est constitué de cellules = éléments figurés du sang, baignant dans un liquide = plasma, qui contient beaucoup d’eau,
des protéines (ex : albumine) et des ions.
Les cellules appartiennent à trois catégories :
• Globule Rouge (GR) = érythrocytes = hématies.
• Globule Blanc = leucocytes :
o monocytes sanguins macrophages
o lymphocytes.
o polynucléaire : neutrophile, éosinophile, basophile.
• Thrombocytes : plaquette sanguine.
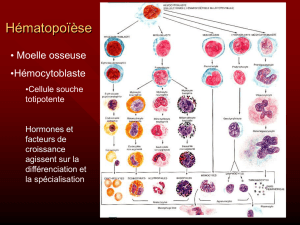
Toutes ces cellules sont formées dans la moelle osseuse, à partir de cellules souches hématopoïétiques totipotentes.
Hématopoïèse = formation de cellule sanguine. Elle est régulée par des molécules = facteur de croissance (ex : EPO =
formation des GR).
II) Exploration du sang des organes hématopoïétiques :
1) Examen des éléments figurés du sang :
• Hémogramme : Analyse quantitative du nombre de cellule sanguine et examen morphologique de celle-ci
mesures effectuées sur les GR : nombre total de GR en millions par mm
3
sang : homme : 4,5 à 6,2, femme : 4 à 6,2
et nouveau né : 5 à 6.
• Hématocrite (centrifugation d’un petit volume de sang lecture directe volume de plasma sur volume de GR) :
homme : 40 à 54 %, femme : 35 à 47 % et nouveau né : 34 à 62 %.
• Taux d’hémoglobine : en g pour 100 mL sang : homme : 13 à 18, femme : 12 à 16 et nouveau né : 14 à 20.
On peut également mesurer le volume globulaire moyen = volume moyen de chaque globule rouge qui est compris entre 85
et 95 µm
3
. Il est fondamental également de connaître le nombre de réticulocyte compris entre 25 000 et 100 000 par mm
3
.
• Exploration des globule blanc : nombre = 4000 à 10 000 par mm
3
.
• Exploration des plaquettes : nombre = 150 000 à 500 000 mm
3
.
A cette analyse du nombre de cellule est associée un examen morphologique des cellules. L’étude morphologique des GB
porte le nom de formule sanguine : Type de GB Nombre absolu par mm
3
Polynucléaire neutrophile (PNN) 1700 – 7000
PN éosinophile 0 – 500
PN basophile 0 – 50
Lymphocyte 1500 – 4000
Monocyte 100 – 1000
On va demander un hémogramme en urgence dans :
• anémie aiguë (baisse du taux d’hémoglobine dans le sang) pâleur, augmentation de la fréquence cardiaque et
respiratoire, coup de barre, etc.
• thrombopénie : manque de plaquette (syndrome hémorragique)
• granulopénie : baisse du taux de PNN, sensible aux infections, fièvre, etc.
2) Exploration de la moelle osseuse :
• myélogramme : il s’agit de l’examen cytologique (examine les cellules) de la moelle osseuse. Prélèvement sternal,
la moelle est aspirée à la seringue puis étalée sur une lame ( sous anesthésie locale avec un trocart).
• Biopsie de moelle : Son but est de réaliser un examen histologique de la moelle (examen des tissus). Dans un
service spécialisé (hématologie), avec un petit trocart permettant de découper un petit fragment osseux dans l’épine
iliaque. Examen complémentaire du myélogramme.
3) Examen des organes lymphoïdes :
Surtout examen des ganglions lymphatiques :
• ponction ganglionnaire entier pour faire l’analyse cytologique.
• biopsie ganglionnaire pour faire l’analyse histologique.
Très souvent les chirurgiens préfèrent prélever le ganglion en entier pour faire l’analyse histologique.
III) Anatomie et physiologie du GR :
2) Structure des GR :
Cellule sans noyau dont le cytoplasme contient essentiellement de l’hémoglobine. Cette protéine contient du fer où se fixe
l’oxygène. Forme en disque bi-concave, cellule déformable pour passer dans les capillaires.
2) Erythropoïèse :
= formation des globules rouges dans la moelle osseuse : cellule souche hématopoïétique érythroblaste réticulocyte
hématie.
Elle dure sept jours, elle est régulée par l’érythropoïétine. Cette construction consomme beaucoup de fer et de vitamine B12
+ folate (= acide folique). La destruction physiologique des GR vieillis se fait dans la moelle osseuse, le foie et un la rate. Ce
sont des macrophages qui assurent cette destruction. Si elle est excessive on parle d’hémolyse. Lors d’une hémolyse

2
anormale on observe une augmentation dans le sang des produits issus de la dégradation des GR (ex : bilirubine). Durée de
vie des GR = 120 jours.
IV) Polynucléaire neutrophile :
Il représente normalement 70 % des GB, avec un noyau polylobé (donc pas polynucléaire), contenant des granulations
cytoplasmiques neutrophile. Fonction :
• cellule douée de mobilité : passe du sang vers les tissus.
• chimiotactisme : cellule attirée sur un foyer infectieux.
• phagocytose : englobement d’une bactérie.
• bactéricide : digestion et destruction de la bactérie.
• Cellule intervenant dans la défense contre les bactéries dans le cadre de l’immunité non spécifique.
V) Polynucléaire éosinophile :
Sa morphologie est caractérisée par des granulations cytoplasmiques orangées. Cellule mobile capable de phagocytose. Rôle
fondamental dans la réaction allergique.
VI) Polynucléaire basophile :
Idem avec granulations cytoplasmiques bleutées. Rôle mal connu, il interviendrait dans la défense contre les parasites.
VII) Monocytes :
Ces cellules sanguines sont capables de migrer dans les tissus sièges de l’infection. Au sein des tissus inflammatoires, elle se
transforment en macrophage tissulaire même rôle et propriétés que les PNN.
VIII) Plaquettes :
Issues de la fragmentation des mégacaryocytes = fragments cellulaires. Elles sont essentiellement impliquées dans le premier
temps de l’hémostase avec la formation du clou plaquettaire. Durée de vie chez l’homme = 10 jours environ.
IX) Produits sanguins labiles :
Ils sont obtenus à partir d’un don de sang total dont la séparation va permettre d’obtenir différents produits. Parmi ces
produits sanguins labiles : concentrés de GR et concentrés de plaquettes.
1) Concentrés de GR :
Les volumes administrés sont d’environ 250 mL/poche. Ces produits sont indiqués dans les tableaux hémorragiques et
d’anémie d’origine centrale (insuffisance de production de GR par la moelle osseuse). C’est plus la tolérance de l’anémie que
le chiffre d’hémoglobine qui impose la transfusion. Généralement, un taux d’hémoglobine < 8g/100 mL de sang doit faire
évoquer la nécessité d’une transfusion.
2) Concentrés plaquettaires :
Ils sont indiqués en cas de thrombopénie (diminution des plaquettes), le but est de maintenir un nombre de plaquette à
20.10
9
/L. Indication des concentrés plaquettaires : hémopathie maligne après chimiothérapie (cancer du sang) et CIVD :
coagulation intra vasculaire disséminée
X) Règles transfusionnelles :
Les GR possèdent au niveau de leur membrane des marqueurs qui définissent les groupes sanguins. Il existe deux systèmes
de groupe sanguin, ABO et rhésus. Outre ces deux systèmes, les GR expriment également d’autres marqueurs.
1) Système A,B,O :
Ou système de glycoprotéines membranaires. C’est la différence au niveau des motifs glucidiques qui sera responsable des
groupes différents. Un sujet possède dans son sérum les anticorps (Ac) dirigés contre les antigènes (Ag) dont il est
dépourvus : Ag sur membrane des GR = groupe sanguin Sérum anticorps
Sujet A : Ag A Ac anti B
Sujet B : Ag B Ac anti A
Sujet AB : Ag A et B Rien
Sujet O : Ag H Ac anti A et B
Groupes sanguins les plus fréquents sont A (45 %) et O (40 %). Les Ac sont dits naturels car ils sont retrouvés dès les
premiers mois de la vie en dehors de toute immunisation. Ces Ac sont réguliers = toujours présents. Les Ag A, B, O sont
responsables d’une spécificité tissulaire. La transfusion de GR doit être iso groupe ou compatible :
Groupes sanguins Concentrés compatibles
O O
A A et O
B B et O
AB A, B, O, AB
2) Système rhésus :
Selon le système rhésus, un sujet peut être positif ou négatif. Les patients rhésus (+) possèdent au niveau de la membrane des
GR l’Ag D. en ce qui concerne les transfusions, il faut des concentrés iso rhésus. Il est particulièrement important qu’un
patient rhésus (-) reçoive du sang rhésus (-).

3
3) Règles de transfusions :
La prescription de concentrés de GR est un acte médical, la réalisation de la transfusion est un acte médicale délégué au para
médical. Elle nécessite :
• un groupage ABO et rhésus sur prélèvements sanguins.
• une recherche d’Ac irrégulier (le RAI : recherche d’agglutinine irrégulière).
• épreuve de compatibilité in vitro avant la transfusion si présence d’Ac irrégulier. Au lit du malade, le contrôle
ultime est obligatoire car il constitue le dernier rempart avant l’erreur de transfusion. Ce contrôle comprend les
points suivants :
o vérification de l’identité du patient + groupe sanguin de celui ci et de la poche.
o vérification de la compatibilité ABO.
4) Accidents transfusionnels :
A) Accidents immédiats :
• Par incompatibilité ABO, on observe un tableau d’hémolyse aiguë intravasculaire. Le patient présente un tableau
évocateur (hypotension, tachycardie, douleur lombaire violente, hyperthermie avec frisson), l’évolution se fait vers
le collapsus cardiovasculaire avec tableau hémorragique, le pronostic est très grave.
• Le syndrome frisson hyperthermie est très fréquent après une transfusion : évolution favorable (aucune risque de
décès). Ce processus est due à la génération d’Ac anti HLA.
• Choc septique, lié parfois à ne septicémie chez le donneur ou à une faute d’asepsie lors du don ou à une mauvaise
conservation du sang. C’est une complication aiguë.
• En cas de transfusion des concentrés plaquettaires, on observe parfois un purpura post transfusionnel aiguë. Ce
processus est due à une destruction des plaquettes du receveur et du donneur par un mécanisme encore mal
expliqué.
B) Accidents différés :
Certaines complications infectieuses sont observables quelques jours à quelques semaines après la transfusion :
• Cytomégalovirus (CMV) : peut être à l’origine d’un syndrome mononucléosique. Infection dangereuse pour les
patients immunodéprimés. On sélectionne les donneurs CMV (-).
• Toxoplasmose : maladie parasitaire également grave chez les immunodéprimés.
• Paludisme possible dans certaines contrées.
C) Complications tardives :
Chez les patients poly transfusés, il existe un risque de surcharge tissulaire en fer = hémochromatose post transfusionnelle.
Pour l’éviter, administration préventive qui chélate (fixe) le fer. Risque infime pour le VHB et VHC ainsi que VIH. Ils sont
systématiquement recherchés dans les dons du sang.
Toutes personnes ayant reçues des produits sanguins labiles doit bénéficier trois mois plus tard d’un contrôle des sérologies
virales (VHB, VHC, VIH).
Le risque de contamination par voie sanguine des agents de types prions n’est pas établit à ce jour. Actuellement, toute
personne ayant des ATCD personnels et ou familiaux de maladies neurologiques non expliquées, ayant reçue de l’hormone
de croissance humaine, et ayant séjournées plus d’un an en Grande Bretagne sont écartées du don du sang.
XI) Aspects médico-légaux de la transfusion sanguine :
Chaque donneur de sang est soumis à un entretien médical, des examens biologiques sont par ailleurs systématiquement
pratiqués. L’éviction définitive concerne les sujets suivants :
• comportement à risque vis à vis du SIDA.
• ATCD d’infarctus du myocarde.
• ATCD d’épilepsie.
• ATCD personnels et ou familiaux de maladies neurologiques non expliquées.
• hépatite B et C, VIH.
L’éviction est temporaire pour les sujets suivants :
• poids < 50 kg.
• femme enceinte et 6 jusqu’à mois après l’accouchement.
• séjour en GB.
• ATCD d’endoscopie de moins de 6 mois.
Toute personne susceptible d’être transfusée doit au préalable être informée. La déclaration des accidents transfusionnels est
obligatoire et se fait à l’établissement français du sang (EFS).
HEMATOLOGIE : Généralités.
1) Moelle osseuse lymphoblastes (précurseurs).
2) Thymus : lieu d’éducation des LT :
• Reconnaissance du soi.
• Reconnaissance des Ag (non soi).
• composé de lymphocytes + cellules de soutien et d’éducation.
• Pathologie : thymome bénin ou malin lymphome.

4
3) Organes lymphoïdes secondaires :
= lieu d’éducation des LB.
Destination finale des lymphocytes éduqués.
Ganglions : formation spécifique le long des axes lymphatiques (relais ganglionnaires) ayant un rôle de filtre.
Ex : cervicaux, spinaux, sus clavier (à gauche CROIZIER), axillaires, inguinaux, articulations moyennes : creux poplité,
coude.
• Réseau de ganglions profonds (péri aortico cab) : du bassin jusqu’au médiastin.
• Circulation lymphatique draine l’ensemble du corps réaction inflammatoire : grossissement, douleur à la
palpation.
• Possibilité de drainer des cellules malignes filtre pour cellules métastatiques.
• Si infection répétées ganglions restent augmentés (si < 1cm non pathologique).
4) Rate :
• Hypocondre gauche. Draine une partie de la circulation digestive : rôle de défense + filtre.
• Muqueuse digestive : première partie d’entrée d’infection.
• Second rôle : renouvellement des différentes cellules sanguines/ destruction des vieilles cellules. Synthèse possible
des cellules sanguines, notamment si leucémie.
Formations muqueuses : 1
ère
ligne de défense :
• Végétation, adénoïdes.
• Agmydales.
• Tractus digestif, respiratoire.
Sang : population lymphoïde qui représente entre 50% (nouveau né, enfant) et 20 % (adulte) des GB, dans ces lymphocytes,
pourcentage de LB et LT (70 %/30 % ou 60 %/40 %).
LYMPHOPOIESE
Au niveau moelle osseuse leucémies.
Au niveau organe lymphoïdes secondaires lymphomes.
Gammapathie : ex : myélome = prolifération de plasmocytes.
Examen :
• Ponction : sous TDM, écho, etc.… : aspect cytologique + immunophénotype dresser la carte d’identité des
marqueurs membranaires d’une population lymphoïde (ce qui détermine la nature cancéreuse et donne la sous
classe de lymphocytes concernés + stade de maturation.
• TDM.
• IRM.
• Imagerie fonctionnelle : PET SCAN on utilise du glucose marqué se fixant là ou il est le plus consommé : cœur,
cerveau, si pathologie : amas de cellules cancéreuses.
HEMATOLOGIE : Hémostase.
Généralités :
• Moyen que possède l’organisme pour arrêter un saignement. Deux phases :
• Hémostases primaire : agrégation plaquettaire.
• Hémostase secondaire : coagulation.
Dans l’organisme, équilibre entre facteurs qui font coaguler et saigner (équilibre dynamique).
Hémostase primaire :
Plaquette :
• Plus petit élément figuré du sang, anucléé, rôle double :
• Brique de construction pour réparer les brèches au niveau des vaisseaux.
• Réservoir de différentes substances de coagulation.
Cellules endothéliales vasculaires :
Toute agression de l’endothélium peut favoriser les phénomènes de thrombose (plaque d’athérome, pathologie lié à la
présence de cathéter dans une veine).

5
Fibrinogène :
Egalement base du caillot (fin de l’hémostase secondaire) + ciment dans l’hémostase primaire.
Facteur de WILLEBRAND :
Consolidation des ponts entre plaquette (hémostase primaire), transporte et protège le facteur 8 de la coagulation (hémostase
secondaire).
Hémostase primaire :
• Adhésion plaquettaire : c’est l’adhésion des plaquettes au niveau de la brèche et des berges.
• Activation des plaquettes qui libère leur contenu dont le facteur de WILLEBRAND, + moyen de fixation
supplémentaire.
• Agrégation plaquettaire : apparition de pont plaquette/plaquette et plaquette/berge en fibrinogène et facteur de
WILLEBRAND. Ainsi les plaquettes ne bougent plus.
• Phénomène rapide, limité dans ses capacités.
Hémostase secondaire :
• Consolide le caillot et fait entrer en jeu tout les facteurs de coagulation présent dans le sang.
• Activation en cascade d’un facteur par l’autre aboutissant à la transformation du fibrinogène en fibrine qui
permettra une réparation tissulaire au long cours.
• Voie intrinsèque : rôle d’amplification.
• TCA : Temps de Céphaline Activée voie intrinsèque et finale (exploration).
• TP : Taux de Prothrombine exploration voie extrinsèque et finale.
• Dosage du fibrinogène.
Hémophile :
Déficit en facteur VIII et IX donc TCA augmentée et TP normal.
+ le TCA monte : + c’est mauvais !
+ le TP baisse : + c’est mauvais !
Régulation : inhibiteur spécifique de la coagulation (antithrombine, protéine CLS).
Deux types d’anticoagulant :
• Héparine (facteur VIII et V, on regarde le TCA).
• Anti vitamine K (facteur VII, on regarde le TP) : fort dosage = mort au rat.
Pathologie :
Signe clinique spécifique de l’hémostase primaire et secondaire.
Déficit de l’hémostase primaire :
• Saignements cutanée et muqueux.
• Au niveau de la peau : purpura (tache rouge ne s’effaçant pas à la pression).
• Saignement de nez, bouche, selle (rectorragie), chez la femme (ménorragie).
• Si coupure, saignement exagéré.
Biologie :
• NFS : dosage des plaquettes.
• Bilan de coagulation : dosage du fibrinogène.
Thrombopénie isolée : confirmer sur tube citraté + myélogramme.
Thrombopénie centrale :
• Infiltration : notamment tumeur.
• Myélodisplasie.
• Médicamenteuse :
o Dépakine :
o Anti coagulant et autres héparines.
Thrombopathies :
• Les plaquettes fonctionnent mal.
• Principale cause : aspirine.
Maladie de WILLEBRAND :
• Déficit de ce facteur (congénital) : symptomatologie de l’hémostase primaire : TCA augmentée, déficit en facteur
VIII.
Thrombopénie périphérique :
 6
7
8
9
6
7
8
9
1
/
9
100%