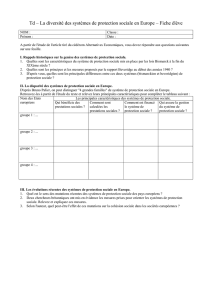
2.2 Dendrogrammes - Manuel de l`évolution biologique

2.2 - Les dendrogrammes
Le dendrogramme est une figure arborescente. Si, dans sa construction, l'on
introduit l'hypothèse que les ressemblances sont le reflet d'une relation de parenté, le
dendrogramme est généalogique ; si l'on introduit celle que les ressemblances
évoluent au cours du temps, le dendrogramme est phylogénétique. Les
dendrogrammes retenus dans cette section mettent en évidence des ressemblances
entre différents taxons.
L'objectif de recherche est souvent double ; dans un premier temps, il s'agit de
mettre en évidence sur un schéma synthétique (le dendrogramme) les relations
généalogiques ou évolutives entre plusieurs taxons ; dans un second temps,
d'apprécier leur degré de divergence. Ce dernier est estimé en fonction soit du temps
qui sépare les taxons, soit des différences génétiques, moléculaires ou autres
accumulées entre ces mêmes taxons.
Les constructions phylogénétiques sont bâties principalement à partir de
l'anatomie comparée, l'ontogénie et la paléontologie.
L'anatomie comparée a pour objectif de rechercher les homologies en utilisant,
par exemple, le principe de subordination des caractères de B. de JUSSIEU ( les
caractères constants sont plus importants que les caractères inconstants), ou encore
celui des connexions de É. GEOFFROY SAINT-HILAIRE (voir la section précédente) : quelles
que soient leur forme, leur taille ou leur fonction, des organes sont reconnus
homologues s'ils possèdent les mêmes connexions avec d'autres organes.
L'ontogénie utilise le principe de récapitulation (loi biogénétique fondamentale)
de E. HAECKEL, mais reformulé par Gareth NELSON (1973) : lorsque l'on peut suivre la
transformation d'un caractère d'un état général vers un état plus spécialisé, le
caractère le plus général est le plus ancien, le moins général est le plus récent, dérivé
du premier. La règle de G. NELSON, qui n'est pas sans rappeler les deux premières
règles de K. E. von BAER, est une hypothèse de travail et non une loi.
La paléontologie fournit des arguments morphologiques, mais aussi des
arguments chronologiques.
À ces données traditionnelles s'ajoutent aujourd'hui celles de la biologie
moléculaire : séquençages des protéines, de l'ADN, de l'ARN, hybridation de l'ADN.
L'unité de base de la construction phylogénétique est très souvent l'espèce,
puisqu'elle est un groupe génétique fermé : l'interfertilité existe uniquement entre ses
103

membres. Mais certains auteurs rejettent cet usage de l'espèce, car leurs travaux
concernent des populations plus que des espèces entières : sous-espèces et espèces
sont alors confondues. Pour éviter l'emploi du mot « espèce », les taxons sont qualifiés
d'unités évolutives (UE), d'unités évolutives hypothétiques (UEH) s’ils sont de pures
constructions fictives, ou encore d’unités taxinomiques opérationnelles (UTO ou OTU
dans la terminologie anglo-saxonne). Les taxons sont parfois de niveau
supraspécifique ; dans ce cas, ils doivent appartenir à une même lignée phylétique
(lignée monophylétique) pour demeurer comparables.
Les arbres sont composés de deux régions : les noeuds où sont placés les
taxons qui sont souvent des UEH, car on ne connaît pas les formes fossiles, et les
branches qui indiquent le degré de parenté des différents taxons. La longueur des
branches est proportionnelle au temps ou bien aux différences entre taxons (fig. 2.15).
À leurs extrémités figurent les taxons terminaux qui sont des UE.
Fig. 2.15
Si l'arbre est enraciné (fig. 2.15-A), la racine représente l'ancêtre commun et il précise
alors les relations évolutives des différents taxons présents. Mais l'arbre est souvent
dépourvu de racines (fig. 2.15-B) : il rend compte uniquement des relations de parenté,
sans que l'on puisse savoir comment l'évolution passe d'un taxon à l'autre. Cependant
un arbre peut être enraciné si on le construit avec un taxon extérieur au groupe, UE
extra-groupe, qui sera la référence pour estimer les degrés de ressemblance entre les
taxons étudiés. Il est nécessaire de connaître précisément les données taxinomiques
104

ou paléontologiques de cette UE extra-groupe ; il faut, en effet, être sûr qu'elle a
divergé bien avant l'ancêtre commun aux UE considérées (fig. 2.15-A). Lorsque l'on
dispose d'un certain nombre d'UE dont on veut établir la parenté, le nombre théorique
d'arbres possibles augmente très rapidement :
- avec n unités (UE) et si l'arbre est enraciné, il y a N1 arbres théoriques, soit :
N1 = (2n - 3) ! : 2 n-2 (n - 2) !
- avec n unités (UE) et si l'arbre n'est pas enraciné, il y a N2 arbres possibles, soit
:
N2 = (2n - 5) ! : 2 n-2 (n - 2) !
Si n = 10, N1 est égal à 35.106 et N2 à 2.106 ; la formule exprime les incertitudes pour
déterminer l'arbre exact parmi plusieurs millions. Un exemple sera donné à propos de
l'émergence de l'Homme moderne à la section 4.4.3 : « Le modèle unirégional ou
monocentrique, discussion ».
Les classifications phylogénétiques utilisent abondamment le critère
d'homologie. Les ressemblances sans lien de parenté sont des homoplasies parmi
lesquelles on distingue les convergences (ressemblances adaptatives) et les
réversions, brusque apparition d'un caractère rappelant un caractère ancestral. Un
caractère ancestral est plésiomorphe ; un caractère dérivé est apomorphe.
Parmi les quatre méthodes principales - phénétique, cladistique, probabiliste et de
compatibilité -, seules seront évoquées les deux premières, car ce sont les plus
fréquentes.
2.2.1 - La méthode phénétique ou numérique
Conçue par Charles MICHENER et Robert SOKAL (1957), elle utilise un nombre
réduit de principes. La construction des arbres phénétiques (phénogrammes) repose
sur les ressemblances observées entre chaque paire d'UE (unité évolutive). Les
ressemblances englobent ici aussi bien les homologies que les homoplasies, les
plésiomorphies (caractères ancestraux) que les apomorphies (caractères dérivés). Les
phénéticiens admettent que les caractères évoluent indépendamment les uns des
autres et qu'ils ont tous le même poids. Plus le nombre de caractères étudiés est
élevé, meilleure sera la classification. Les ressemblances entre UE sont souvent
estimées par l'emploi d'une matrice de similitude (voir ci-dessous la méthode UPGMA).
Peter SNEATH et R. SOKAL précisent que les phénogrammes ne sont pas a priori
phylogénétiques ; en fait, ces arbres doivent être reconnus seulement comme
105

phénétiques. Le phénogramme n’a pas de racines, car il montre les relations
morphologiques qui rapprochent ou qui éloignent plusieurs unités taxinomiques. Mais
la notion d'évolution (phylogénie) peut se déduire des phénogrammes à condition
d'introduire dans la construction des hypothèses évolutives telle que l'horloge
moléculaire, par exemple.
On distingue trois méthodes phénétiques majeures :
- La méthode d'ajustement. L'arbre non enraciné choisi est celui dont les longueurs
des branches expliquent le mieux les ressemblances des UE ; l'introduction de
certains critères permet de déterminer les longueurs.
- La méthode de parcimonie. L’objectif, qui rappelle celui de la méthode cladistique,
est d’obtenir un arbre non enraciné le plus court possible avec une minimisation des
homoplasies.
- Les méthodes d'agglomération. La classification est hiérarchique car les UE sont
classées en fonction de leurs ressemblances. La méthode agglomérative dite UPGMA,
abréviation anglaise de Unweighted Pair Group Method with Arlthmetic Mean, a été
très employée par les phénéticiens pour traiter les données moléculaires (séquençage,
etc.). Si la notion de l'horloge moléculaire est admise ; elle implique que le taux de
mutations et la vitesse d'évolution d'un caractère donné sont constants. Par
conséquent, les longueurs des branches de l'arbre peuvent être proportionnelles au
temps. En revanche, la méthode dite « du plus proche voisin » (neighbor joining), qui
se développe rapidement, n'utilise pas le postulat de l'horloge moléculaire. Les paires
d'UE sont regroupées d'après leurs ressemblances, de telle sorte que l'arbre non
enraciné construit soit le plus court possible.
Les résultats qui proviennent de l'étude de séquençage sont exprimés sous
forme de chiffres qu’on soumet à un certain nombre de manipulations : ce tableau
devient alors une matrice. La manière la plus simple consiste à quantifier les
différences entre chaînes peptidiques (protéines) et nucléotidiques (ADN ou ARN).
Tout calcul doit être précédé de l'alignement des séquences, même lorsque les
chaînes sont de longueur identique. La figure 2.16 donne un exemple de matrice
simple où sont reportés les nombres d'acides aminés différents de la chaîne α de
l'hémoglobine, chez quelques Vertébrés ; les séquences sont comparées deux à deux.
106

Le nombre de différences correspond à une distance génétique : deux espèces seront
d'autant plus proches que leurs différences seront faibles.
La figure 2.17 montre un exemple simple de calculs permettant la construction d'un
phénogramme avec les transformations successives subies par la matrice.
107
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
1
/
24
100%