Facteurs étiologiques généraux de la pathologie pulpodentinaire

Facteurs étiologiques généraux
de la pathologie pulpodentinaire
B. Alliot-Licht
V. Armengol
S. Dajean-Trutaud
D. Marion Résumé. –Certaines pathologies générales peuvent avoir des conséquences sur la pulpe et la dentine. Au
niveau pulpaire, elles peuvent provoquer des nécroses, des calcifications et des métaplasies. Au niveau
dentinaire, un excès ou un défaut de dentinogenèse et/ou de minéralisation sont observés. Une revue de la
littérature récente permet d’évoquer les diverses pathologies pulpodentinaires et les mécanismes
pathogéniques en cause induits par le diabète non contrôlé, l’athérosclérose, l’hyperbilirubinémie, les
pathologies rénales chroniques, l’oxalose, l’anachorèse, les troubles de la parathyroïde, l’hypervitaminose D,
la sclérodermie, certaines tumeurs malignes, des maladies génétiques comme les rachitismes héréditaires,
l’hypophosphatasie, l’ostéogenèse imparfaite et divers syndromes héréditaires rares, les carences diététiques
en ions et en vitamines, les intoxications mercurielles, l’infection fongique à Candida, le zona, la lèpre, ainsi
que divers traitements généraux (glucocorticoïdes, tétracyclines, suppléments fluorés et traitements
anticancéreux).
©2004 Elsevier SAS. Tous droits réservés.
Mots-clés : Pathologies générales ; Syndromes héréditaires ; Pulpopathies ; Pulpolithe ; Dentinogenèse ;
Défaut dentinaire
Introduction
Les facteurs locaux responsables des pathologies pulpaires, ainsi que
des altérations dentinaires, sont généralement bien connus des
cliniciens. En revanche, les étiologies générales susceptibles d’être
impliquées dans ces phénomènes leur sont beaucoup moins
familières et ne sont donc que rarement évoquées dans le diagnostic
étiologique des pulpopathies.
Pourtant, les progrès de la médecine ces dernières décennies
permettent de maintenir en vie des malades, en particulier des
enfants, atteints de pathologies autrefois fatales. Ces patients, malgré
des handicaps parfois lourds, ont maintenant besoin de soins
dentaires et il importe de connaître les répercussions de ces
pathologies sur leur état buccodentaire, et notamment pulpaire, afin
d’adapter nos traitements.
Si de nombreux travaux de recherches cliniques et fondamentales
ont porté sur l’influence des lésions dentaires sur les pathologies
générales (infections focales, endocardites bactériennes, rôles des
parodontopathies sur l’évolution des maladies systémiques…),
l’intérêt des chercheurs est nettement moins axé sur la manière dont
une maladie générale peut avoir une répercussion sur la santé
pulpodentinaire. Une revue de la littérature récente permet de faire
la part de ce qui relève dans ce domaine des lieux communs non
prouvés scientifiquement et de ce qui peut être étayé et expliqué par
des études cliniques ou des protocoles expérimentaux
reproductibles. Nous limiterons ce travail sur les étiologies générales
des pathologies pulpodentinaires aux seuls facteurs endogènes,
c’est-à-dire provenant directement de pathologies générales propres
aux patients ou consécutives à leurs traitements, en excluant donc
tout ce qui est en rapport avec des étiologies exogènes, liées à
l’environnement (barotraumatismes, variations thermiques,
irradiations…). De même, nous n’évoquerons pas ce qui résulte de
phénomènes physiologiques, tels que le vieillissement, ainsi que de
pathologies héréditaires n’affectant que les seuls tissus dentaires.
Pathologies pulpodentinaires
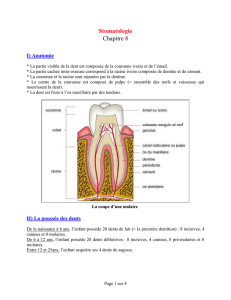
Le tissu pulpaire est un tissu conjonctif spécialisé, fibreux, qui assure
les fonctions dentinogénétiques, nutritives, sensorielles et de défense
de la dent. La palissade d’odontoblastes située en périphérie de la
pulpe contrôle les mouvements et la composition ionique du fluide
présent dans les tubuli dentinaires et intervient dans la sécrétion et
le transport des composants nécessaires à la dentinogenèse. La
formation de la dentine qui est sous le contrôle de nombreux
facteurs systémiques (hormones et vitamines) et locaux (facteurs de
croissance) passe par la synthèse d’une couche de prédentine
essentiellement composée de collagène de type I qui se minéralise
secondairement. La minéralisation de la prédentine dépend de la
concentration locale en phosphates, en calcium et en protéines
spécifiques. Une pathologie générale qui provoque une perturbation
des ions, des hormones, des vitamines et de la matrice impliqués
dans la dentinogenèse peut donc entraîner un manque ou un excès
et/ou des malformations de la dentine.
La vascularisation pulpaire est terminale, il n’existe pas de
circulation collatérale et l’ensemble du paquet vasculonerveux
pénètre dans la pulpe par le foramen apical. Une pathologie
vasculaire d’ordre général pourra donc avoir des répercussions
dramatiques sur le tissu pulpaire et par voie de conséquence, sur la
dentinogenèse.
Toute irritation de l’organe pulpodentinaire entraîne inévitablement
une inflammation pulpaire aiguë ou chronique dont l’évolution
B. Alliot-Licht (Maître de conférences des Universités, praticien hospitalier)
V. Armengol (Maître de conférences des Universités, praticien hospitalier)
S. Dajean-Trutaud (Maître de conférences des Universités, praticien hospitalier)
D. Marion (Maître de conférences des Universités, praticien hospitalier)
Adresse e-mail : [email protected]
Faculté de chirurgie dentaire, 1, place Alexis-Ricordeau, B.P. 84215, 44042 Nantes cedex 1, France.
Encyclopédie Médico-Chirurgicale 23-009-A-10
(2004)
23-009-A-10

dépend de l’intensitéet de la duréedel’agression, mais également
de l’état préalable de la pulpe. L’atteinte pulpaire peut évoluer en
fonction de facteurs étiologiques généraux (maladies, traitements)
qui provoquent soit une irritation pulpaire en tant que telle, soit une
diminution des capacitésdedéfense ou de régénération de la pulpe.
De plus, sans étiologie locale associée, la pulpe dentaire peut subir
chez des patients atteints de pathologies générales, une
dégénérescence de différentes natures : une fibrose pulpaire
caractérisée par un nombre élevéde fibroblastes et une synthèse
exagéréedefibres matricielles, une dégénérescence calcique avec des
calcifications (pulpolithes) plus ou moins importantes dans la pulpe
camérale et radiculaire, responsables d’un rétrécissement de l’espace
canalaire, et une dégénérescence nécrotique aboutissant àune
nécrose de la pulpe, accompagnéeengénéral de complications
périapicales. Une pathologie générale peut également entraîner une
métaplasie pulpaire àl’origine de résorption dentinaire interne.
Enfin, des mécanismes de défense inappropriés (sur des dents ne
présentant ni carie, ni traumatisme) peuvent se produire dans la
pulpe (dentine tertiaire) et au niveau de la dentine (formation de
dentine sclérotique par fermeture des tubuli).
[1]
Pathologies générales et leurs
conséquences pulpodentinaires
DIABÈTE
Le diabète sucréest une maladie qui touche le métabolisme des
hydrates de carbone et des lipides. Cette pathologie est causée par
un trouble de la sécrétion ou de la fonction d’une hormone,
l’insuline, provoquant une augmentation de la glycémie. Il existe
deux types de diabètes. Le diabète de type I, associéàune anomalie
ou àl’absence des cellules qui produisent l’insuline, cellules bêta
localisées dans les îlots de Langerhans du pancréas, apparaît chez
l’enfant ou l’adolescent. Le diabète de type I est dit
insulinodépendant car les malades ont besoin d’un apport d’insuline
exogène pour survivre. En absence d’un traitement régulier, les
malades peuvent développer une acidose grave avec comas et
diverses lésions dégénératives. Le diabète de type II, appelénon
insulinodépendant ou diabète gras est plus courant. Il est dûàune
diminution de la fonction des cellules bêta ou àune résistance à
l’insuline. Les malades atteints de diabète de type II ne peuvent pas
développer d’acidose grave et leur traitement consiste àstimuler la
sécrétion d’insuline. Classiquement, le diabète de type II apparaîtà
partir de 50 ans mais un surpoids peut provoquer un diabète dès
l’enfance.
Quel que soit le type de diabète, l’excès de glucose dans le sang
provoque de nombreux troubles vasculaires. Tous les vaisseaux sont
atteints, de l’aorte aux fins capillaires. L’hyperglycémie provoquée
par un diabète non contrôléentraîne une déshydratation cellulaire
et la formation excessive d’hémoglobine glycosylée(HbA
1c
) moins
efficace dans le transport de l’oxygène et responsable de
l’épaississement de la membrane basale des capillaires. Cette
modification a des conséquences sur la réponse immunitaire. Elle
diminue la réponse leucocytaire et entrave le rôle antibactérien des
polymorphonucléaires (PMN). Les diabétiques ne sont pas plus
sujets aux infections bactériennes mais présentent une plus grande
probabilitéde développer une infection grave en raison de cette
diminution de la réponse immunitaire mais aussi de l’augmentation
de la teneur en sucre dans la région pathologique qui peut accélérer
la multiplication bactérienne.
[2]
Contrairement aux maladies parodontales, les conséquences du
diabète non contrôlésur la pulpe dentaire ont étépeu observées.
Seules deux études effectuées dans les années 1960
[3, 4]
sur l’état
histopathologique des pulpes dentaires de patients diabétiques
révèlent la présence de calcifications pulpaires en forme de faucille
[3]
ou amorphes.
[4]
En revanche, seule l’étude de Russell
[3]
fait état de
troubles vasculaires et d’épaississement des membranes basales des
capillaires pulpaires.
Chez les patients diabétiques non contrôlés, les dommages
vasculaires et l’inhibition de l’activitéantibactérienne des PMN sont
considérés comme des facteurs aggravants du risque d’infection
pulpaire. Une augmentation du nombre des lésions périapicales est
observée chez les patients dont le diabète n’est pas contrôlé.
[5]
En
revanche, lorsque la glycémie est équilibrée, la prévalence de lésions
périapicales est identique àcelle observée sur les patients non
diabétiques.
[2]
De plus, des résultats récents semblent montrer une
augmentation de la prévalence d’agents microbiens endodontiques
dans la pulpe nécrotique de patients diabétiques.
[5]
Bender
[2]
décrit le cas d’une patiente de 32 ans qui présente des
douleurs dentaires bilatérales, sans rapport avec des dents cariées et
dont huit dents ne répondent pas positivement aux tests de vitalité
pulpaire. Le diabète de cette patiente a étédiagnostiquéet donc
traitéàla suite de cette première visite. Six mois après, l’examen
radiologique révèle la présence de lésions périapicales sur les dents
qui ne répondaient pas aux tests de vitalité. Ces cas d’« odontalgie
diabétique »qui apparaissent en absence de carie sont attribués aux
processus d’anachorèse ou aux troubles circulatoires liés au diabète
non contrôlé. Cependant, des études plus poussées sont nécessaires
pour mieux comprendre ces mécanismes.
[2]
ATHÉROSCLÉROSE
L’athérosclérose est une pathologie dans laquelle l’obstruction de la
lumière des artérioles par des plaques athéromateuses (dépôts
lipidiques) provoque une ischémie et une dégénérescence tissulaire.
Les patients souffrant d’athérosclérose coronaire présentent un plus
grand nombre de dents avec des calcifications pulpaires que des
patients sains.
[6]
Bernick et al.
[7]
ont aussi décrit des altérations
pulpaires athérosclérosiques (calcification, nécrose), mais ces études
portent sur des patients âgésde40à70 ans et il est connu que l’âge
provoque des modifications considérables au niveau des vaisseaux
pulpaires. L’effet de l’athérosclérose seule sur la pulpe dentaire n’a
pas étéconfirméchez l’animal. Oguntebi et al.
[8]
ont mis en évidence
un rétrécissement des artérioles pulpaires sur des porcs miniatures
présentant une hypercholestérolémie induite. Des plaques
d’athéromes ont étéobservées mais la forte concentration de
cholestérol n’a jamais provoquéd’obstruction vasculaire complète
au sein de la pulpe. De même, aucun changement dégénératif
(nécrose ou calcification) n’aétédécelédans les tissus pulpaires
examinés.
[8]
L’absence d’altération pulpaire a étéconfirmée sur des
singes sur lesquels une arthérosclérose a étéinduite
expérimentalement.
[9]
Il semble donc que les artérioles pulpaires ne
soient pas propices au dépôt athéromateux.
HYPERBILIRUBINÉMIE
La bilirubine est le pigment jaune rougeâtre présent entre autres
dans la bile et le sérum. Elle provient de la dégradation de
l’hémoglobine par perte du fer. Dans les cas d’anémie hémolytique
précoce àbilirubine indirecte dont l’origine peut être une
incompatibilitéde types sanguins Rhésus fœtomaternelle ABO
(érythroblastose fœtale),
[10]
ou dans les pathologies provoquant des
dysfonctions hépatiques ou biliaires ainsi que dans les pathologies
qui déclenchent une hémolyse, l’excès de bilirubine provoque des
colorations des dents temporaires. La bilirubine pénètre dans la
dentine donnant une couleur jaune, brune, grise ou bleutée. Les
dents prennent une couleur verdâtre lorsque la bilirubine absorbe la
lumière bleue et s’oxyde en biliverdine. Ces colorations
particulièrement visibles au niveau radiculaire s’estompent avec
l’âge.
[1, 11, 12]
23-009-A-10 Facteurs étiologiques généraux de la pathologie pulpodentinaire Odontologie
2

PATHOLOGIE RÉNALE CHRONIQUE
Lors des pathologies rénales, quelle qu’en soit l’origine, primaire ou
secondaire àun diabète, àune hypertension ou àune
glomérulonéphrite, le processus pathologique conduit àune
détérioration puis àune destruction des néphrons. Une des
conséquences de la perte de la fonction des néphrons est une
diminution de la filtration des glomérules rénaux, provoquant une
augmentation de la concentration en phosphate dans le sérum.
L’excès de phosphate entraîne une incorporation du calcium dans le
tissu osseux par régulation homéostasique. Il en résulte une
diminution de la concentration du calcium dans le sérum qui
stimule la glande parathyroïde. Un autre effet des pathologies
rénales est l’impossibilitéde produire la forme active de la vitamine
D (1,25-dihydroxyvitamine D
3
), ce qui entraîne une diminution de
l’absorption intestinale du calcium et une sécrétion continue
d’hormone parathyroïdienne. Des troubles osseux (ostéodystrophie
rénale) de type ostéomalacie, ostéite fibreuse généraliséeou
ostéosclérose apparaissent alors. Après une greffe de rein, toutes les
valeurs redeviennent normales.
[13]
Au niveau dentaire, les patients
souffrant de pathologies rénales chroniques ayant subi une
transplantation rénale présentent des calcifications pulpaires et un
rétrécissement de la chambre pulpaire par excès de dentinogenèse
secondaire.
[14, 15, 16]
Il est possible que le traitement
immunosuppresseur àbase de glucocorticoïdes administréaux
patients ayant subi une greffe rénale soit responsable du
rétrécissement de la chambre pulpaire.
[15]
Mais il est observéun
épaississement de la prédentine sur des patients insuffisants rénaux
chroniques n’ayant reçu aucun traitement àbase de corticoïdes.
[14,
17]
L’excès de production de prédentine irrégulière est aussi expliqué
par l’hypophosphatémie et le rachitisme dus àla déficience en
vitamine D
3
ou par l’hyperparathyroïdisme observélors d’une
insuffisance rénale chronique.
[14, 17]
Les affections rénales peuvent être associées àune ostéomalacie due
àune contamination par l’aluminium dans l’eau de dialyse ou àla
conséquence d’un traitement àbase de sel d’aluminium prescrit
pour réguler le phosphate. Quoi qu’il en soit, l’aluminium
s’accumule au front de minéralisation des tissus calcifiésoùil
pourrait inhiber la minéralisation. De plus, une oxalose secondaire
en association avec une intoxication en aluminium serait responsable
de la perte des dents àla suite de résorptions radiculaires internes
et externes.
[18]
HYPEROXALURIE ET OXALOSE
L’hyperoxalurie, qu’elle soit primaire (maladie héréditaire rare dont
le mode de transmission est autosomique récessif) ou secondaire à
une pathologie rénale, provoque un dépôt de cristaux d’oxalate de
calcium appeléoxalose dans les tissus conjonctifs extrarénaux. La
forte concentration en oxalate plasmatique et les altérations de la
perméabilitévasculaire seraient responsables de la formation de ces
cristaux.
[19]
L’hyperoxalurie primaire est caractérisée par des
altérations rénales graves (néphrolithiases, néphrocalcinose et
précipitations de cristaux d’oxalate dans les reins) nécessitant des
dialyses et àterme une transplantation rénale. Chez des patients qui
présentent une hyperoxalurie primaire, des manifestations dentaires
comme des douleurs et des résorptions apparaissent, mais
uniquement avec une pathologie rénale chronique associée.
[18]
Hedemark
[20]
a montré, sur une patiente de 25 ans qui souffrait
d’une hyperoxalurie primaire, la présence de cristaux d’oxalate dans
la pulpe dentaire fibrosée. Cette patiente ne présentait aucune
pathologie dentaire jusqu’à l’âge de 18 ans, puis un traitement
orthodontique a étémis en œuvre et sans que ce traitement puisse
être incriminé, de nombreuses résorptions dentaires sont apparues.
Pendant le traitement orthodontique, cette patiente a subi deux
greffes rénales puis une greffe rein-foie.
[20]
Dans d’autres cas
similaires, la présence de cristaux d’oxalate a étédécrite dans la
pulpe dentaire mais aussi au niveau de la prédentineetdeladentine
tertiaire.
[21]
Des zones de résorption interne sont comblées par de
l’ostéodentine qui s’étend dans la pulpe et qui contient des cristaux
d’oxalate. Enfin, des pulpolithes formés autours des cristaux
d’oxalate de la pulpe fusionnésavecl’ostéodentine provoquent de
larges calcifications intrapulpaires.
[22]
L’étiopathogénie de ces lésions pulpaires pourrait être
l’hypovitaminose d’origine rénale qui induit àson tour un déficit en
ostéopontine, glycoprotéine phosphorylée connue pour inhiber la
formation de cristaux d’oxalate de calcium.
[23]
La réaction
granulomateuse inflammatoire induite par les cristaux d’oxalate de
calcium dans la pulpe serait àl’origine des résorptions internes des
dents
[18, 21]
et des douleurs pulpaires.
[21]
ANACHORÈSE
L’anachorèse est la localisation et la fixation, dans des zones
inflammatoires, de micro-organismes provenant d’une autre source
et véhiculés par voie sanguine.
[2, 24]
Ilaétéprouvéque les réactions
inflammatoires causées par les soins dentaires sont capables d’attirer
vers la pulpe, par voie hématogène, des micro-organismes issus
d’une pathologie infectieuse éloignée de la dent.
[2]
Chez le chien, 24 heures après une injection intraveineuse d’une
suspension de streptocoques a, aucun micro-organisme n’est décelé
dans les pulpes contrôles non enflammées ; en revanche, dans toutes
les pulpes ayant subi un coiffage direct àl’hydroxyde de calcium et
présentant donc une inflammation, la pulpe est infectée par des
micro-organismes identiques àceux qui ont étéinjectés par voie
sanguine. Les auteurs en concluent que les pulpes non
inflammatoires n’attirent pas les bactéries par voie anachorétique et
que l’action chimique de l’hydroxyde de calcium peut être
considérée comme un facteur favorisant l’infiltration vers la pulpe
de bactéries issues du sang.
[24]
Le processus d’anachorèse explique comment la pulpe dentaire peut
développer des nécroses ou des pulpites «a retro »sans carie et sans
infiltration bactérienne àpartir d’une infection àdistance sur des
patients souffrant de pathologie générale comme le diabète.
[2]
TROUBLES DE LA PARATHYROÏDE
La parathormone (PTH) est une hormone qui agit àdeux niveaux :
le tissu osseux oùelle régule le remodelage osseux, et les reins où
elle augmente la sécrétion de phosphate et inhibe la sécrétion de
calcium. De plus, la PTH catalyse l’hydroxylation de la vitamine D
qui àson tour contrôle la minéralisation et l’absorption intestinale
du calcium.
[25]
¶Hyperparathyroïdisme
Une hyperparathyroïdie primaire est le plus souvent engendrée par
un adénome bénin de la glande parathyroïde. L’hyperparathyroïdie
secondaire àune anomalie rénale mobilise àla fois le calcium et le
phosphate àpartir du tissu osseux.
Sur une patiente de 18 ans, une calcification complète de la pulpe a
étéobservée sur deux dents ne présentant que de petites lésions
carieuses.
[26]
L’hyperparathyroïdisme a étéévoquépour expliquer
l’apparition de ces calcifications pulpaires ; cependant, dans ce cas,
toutes les valeurs biologiques étant normales, l’origine de ces
calcifications semble idiopathique.
De plus, de nombreuses recherches ont montréque les hormones
qui régulent la calcémie jouent un rôle important dans la
minéralisation de la dentine. Chez la souris, une forte dose de PTH
n’a pas d’effet sur la différenciation des cellules mésenchymateuses
en préodontoblastes mais interfère avec la cytodifférenciation des
préodontoblastes en odontoblastes et inhibe la formation de
prédentine.
[27]
Odontologie Facteurs étiologiques généraux de la pathologie pulpodentinaire 23-009-A-10
3

¶Hypoparathyroïdisme
Les dents de patients atteints d’hypoparathyroïdie, de candidose et
de retard mental ont étéexaminées. Tous les malades présentaient
une concentration sérique en calcium très basse et un taux en
phosphate trèsélevé. Au niveau dentinopulpaire, des cavités
pulpaires larges dues àune hypoplasie résultant d’un arrêtdela
dentinogenèse sont décrites ainsi que l’obstruction des chambres
pulpaires par de l’ostéodentine. Cependant, il est difficile de savoir
si ces anomalies sont dues directement àl’hypoparathyroïdisme.
[25]
Un défaut de minéralisation par la formation de dentine
interglobulaire au niveau radiculaire est rapportédans les cas de
pseudohypoparathyroïdisme, maladie due àl’absence de réponse
des cellules osseuses et rénales àla PTH et dans les cas
d’hypoparathyroïdisme idiopathique oùles anomalies dentinaires
sont associées àdes infections pulpaires répétées liées àCandida.
[11]
HYPERVITAMINOSE D
La fonction biologique principale de la 1,25-dihydroxyvitamine D
est de maintenir une concentration normale en calcium et en
phosphate dans le sérum afind’assurer les fonctions cellulaires
essentielles et de permettre la minéralisation des tissus durs. Les
besoins en vitamine D sont en général fournis par l’alimentation et
l’exposition aux rayonnements solaires. Giunta
[28]
décrit un cas rare
d’hypervitaminose D due àun excèsd’absorption de lait enrichi en
vitamine D, chez un enfant de 7 ans qui présente de nombreux
troubles digestifs et des calcifications ectopiques. L’excèsde
vitamine D a entraînéune hypercalcémie marquée et prolongée due
àune accélération de l’absorption intestinale et àune résorption du
tissu osseux. Au niveau pulpaire, il est notél’apparition de
calcifications.
[28]
De même, des ponts radio-opaques sont observés
au niveau des chambres pulpaires de toutes les incisives
permanentes maxillaires et mandibulaires dans un cas
d’hypercalcémie iatrogène d’un enfant atteint d’ostéodystrophie
rénale et traitépar de trop fortes doses de vitamine D.
[29]
Chez
l’animal, en plus des calcifications pulpaires, une déformation des
racines, un vieillissement prématurédu complexe pulpodentinaire
et la formation d’ostéodentine ont étémis en évidence.
[30]
Pour
mieux comprendre les mécanismes responsables de ces excèsde
minéralisation dentaire, il a étémontrésur des cultures de cellules
pulpaires que la vitamine D augmente la synthèse d’ostéopontine
associéeàla formation de dentine de réparation et àl’apparition de
calcifications pulpaires.
[23]
SCLÉRODERMIE
La sclérodermie est une maladie des tissus mésenchymateux
caractérisée par une prolifération excessive de collagène dans les
tissus sous-cutanés. Cette pathologie, plus fréquente chez la femme,
se développe sur un mode chronique. Plusieurs étiologies de la
sclérodermie ont étéproposées, des facteurs neurotrophiques, une
anomalie fonctionnelle de la glande thyroïde mais aussi certaines
conditions atmosphériques, des intoxications médicamenteuses, des
infections aiguës ou des réactions allergiques. Au niveau dentaire,
la fermeture localisée des tubuli dentinaires entraîne une diminution
du nombre des tubuli principalement àproximitéde la chambre
pulpaire. Une étude en microanalyse montre de nombreuses
modifications de la composition minérale de la dentine avec une
augmentation du taux de phosphate, la disparition du magnésium
et une modification du rapport calcium-phosphate.
[31]
TUMEURS MALIGNES
¶Lymphome de Burkitt
Le lymphome de Burkitt est un lymphome malin constituépar la
prolifération des cellules souches lymphoïdes et frappant les
adolescents de certaines régions d’Afrique, mais il existe une forme
européenne. Le virus Epstein-Barr (agent étiologique de la
mononucléose infectieuse) serait co-responsable de ce lymphome qui
se développe dans les maxillaires en envahissant les tissus mous.
Dèsledébut de la maladie, les études histologiques montrent que
les papilles dentaires des dents voisines sont envahies par le
processus lymphomateux. La tumeur pénètre dans la papille
dentaire par l’apex provisoire et progressivement les cellules
lymphoïdes remplacent les cellules pulpaires.
[32]
Face aux
traitements chimiothérapiques, le lymphome de Burkitt régresse
rapidement et le tissu osseux se régénère autour des dents mais
l’examen histologique des dents après traitement montre une
dentine irrégulière avec une démarcation entre la dentine normale,
formée avant la maladie et la dentine formée pendant la maladie.
Cette démarcation peut être une bande étroite de dentine moins
minéralisée ressemblant àde la prédentine. Dans les dents en cours
d’évolution pendant le lymphome, après le traitement, dans la
majoritédes cas, le diaphragme épithélial est préservé,la
dentinogenèse radiculaire se poursuit normalement et une pulpe
saine est observable. Si le diaphragme épithélial est détruit, une
fibrose pulpaire se produit. La formation de dentine normale après
le traitement est le signe de la présence d’odontoblastes sains qui
ont pu remplacer les odontoblastes détruits lors du processus
métaplasique. Une déformation de la racine après traitement et
guérison du lymphome est souvent observée et peut être considérée
comme une preuve indirecte de l’envahissement de la papille par la
tumeur.
[32]
¶Infiltration métastatique
Un cas de métastase maxillaire envahissant la pulpe dentaire a été
récemment décrit
[33]
àpartir d’un médulloblastome, la plus
fréquente des tumeurs cérébrales chez l’enfant, représentant 15 à
20 % des tumeurs pédiatriques. Les métastases extracérébrales de
ces tumeurs sont rares. Dans le cas de ce jeune enfant, la
radiographie panoramique permet d’observer la présence d’une
image radioclaire autour de certaines dents. L’examen histologique
révèle une importante infiltration de la pulpe dentaire des dents
temporaires et de la papille des dents définitives en formation par
des cellules tumorales.
La destruction de la pulpe par des métastases périapicales dans le
cas d’une tumeur maligne a étédécrite sur une femme de 45 ans.
[34]
Une biopsie de la pulpe et du périapex des dents présentant des
images radioclaires périapicales a révéléque la lésion périapicale
contenait des cellules métastatiques alors que les fragments
pulpaires présentaient des calcifications, des cellules pycnotiques et
n’avaient plus de vascularisation. Contrairement au cas précédent,
les modifications néoplasiques sont restées confinées dans les
maxillaires et n’ont pas envahi la pulpe dentaire. La destruction de
la pulpe serait la conséquence de l’arrêt de la circulation sanguine
pulpaire dûàla prolifération des cellules tumorales au niveau
périapical.
MALADIES GÉNÉTIQUES
¶Rachitisme héréditaire
Rachitisme hypophosphatémique vitamine D-résistant (HVDRR)
Il existe deux types d’hypophosphatémie héréditaire appelée aussi
rachitisme familial résistant àla vitamine D : l’hypophosphatémie
liéeàl’X (HLX), cas le plus courant de rachitisme héréditaire dans
lequel le trouble de la réabsorption du phosphate au niveau rénal
provoque une hypophosphatémie avec un rachitisme sévère, une
ostéomalacie et une synthèse anormale de dihydroxyvitamine D
3
et,
d’autre part, la maladie osseuse hypophosphatémiante autosomique
23-009-A-10 Facteurs étiologiques généraux de la pathologie pulpodentinaire Odontologie
4

dominante (HBD), pathologie héréditaire dans laquelle le trouble de
réabsorption rénale, différent de celui de l’HLX, entraîne des
anomalies osseuses mais pas de rachitisme uniforme bien que le
niveau de phosphate dans le sérum soit aussi faible.
[35]
Les signes dentaires de ces deux syndromes sont un émail normal
mais fin, une cavitépulpaire élargie avec des cornes pulpaires qui
s’étendent jusqu’à la jonction émail/dentine et un taurodontisme.
La taille anormale de la pulpe est la conséquence d’une dysplasie de
la dentinogenèse secondaire et/ou d’une déficience de la
minéralisation (dentine interglobulaire).
[36, 37, 38, 39, 40]
Un excès de zinc
dans les espaces interglobulaires pourrait expliquer ce trouble de la
minéralisation dentinaire.
[41, 42]
En effet, contrairement au tissu
osseux, le déficit en phosphate ne semble pas être la cause principale
des défauts de minéralisation dentinaires, tandis que le zinc, connu
pour inhiber la minéralisation dentinaire et observéen excès dans
les espaces interglobulaires sur un modèle de souris HLX, jouerait
un rôle déterminant.
[41]
Les malades atteints d’HLX et d’HBD présentent tous des anomalies
dentaires mais àdes degrésdifférents, alors que l’hypophosphatémie
est similaire. De plus, les lésions pulpodentinaires sont observables
même lorsque les malades ont un traitement dèsl’enfance qui
permet de restaurer le taux de phosphate sérique àdes valeurs
normales.
[43]
Ilaétédémontréque les anomalies dentaires observées
dans les cas d’HLX sont la conséquence de deux processus séparés,
l’un qui est dûàl’hypophosphatémie d’origine rénale, l’autre
affectant directement la dentinogenèse secondaire.
[36]
Une fois
induite génétiquement, la dentinogenèse secondaire est continue,
lente et régulière et ne semble pas dépendre de la concentration en
phosphate.
[36, 44]
Des nécroses pulpaires (dans 40 % des cas d’HBD, 50 % des HLX
chez la femme et 100 % des HLX chez l’homme) et des abcèsau
niveau de dents temporaires et définitives non cariées sont décrits
sans que la cause de ces abcès soit clairement définie.
[38, 39]
Au
niveau des dents temporaires, la faible épaisseur d’émail puis
l’envahissement bactérien de la dentine hypominéraliséeou
l’attrition amélaire entraînant une exposition pulpaire au niveau des
cornes pulpaires effilées pourraient expliquer la nécrose pulpaire
[42]
alors que sur les dents définitives, les abcès sont inexpliqués.
[38]
La
progression des micro-organismes dans les défauts microscopiques
de l’émail et de la dentine semble une hypothèse probable.
[42]
Bien
que ces abcès dits «spontanés»apparaissent dèsl’âge de 2 ans,
l’intérêt de pulpotomies prophylactiques chez les jeunes patients
atteints de rachitisme résistant àla vitamine D a étédiscuté
[37]
et
depuis 2002, ce traitement n’est plus recommandé.
[39]
Dans la pathologie liéeàl’X, les anomalies dentaires sont plus
accentuées chez les hommes que chez les femmes (ceci est dûà
l’atténuation du phénotype par l’allèle sain).
[35, 36, 37, 45]
Rachitisme vitamine D-dépendant (VDDRI et VDDRII)
La bioactivation de la vitamine D requiert l’activitéenzymatique de
la 1a-hydroxylase rénale. La déficience ou l’absence de cette enzyme
est provoquée par une maladie autosomale récessive rare appelée
rachitisme vitamine D-dépendant de type I (VDDRI ou rachitisme
vitamine D-pseudodéficient). Le VDDRII ou rachitisme vitamine
D-dépendant de type II (aussi nommérachitisme hypocalcémique
vitamine D-résistant) est une autre forme de rachitisme à
transmission autosomique récessive causée par une anomalie du
récepteur àla vitamine D et donc résistante au traitement
vitaminique. Les signes cliniques du syndrome du VDDRI sont
nombreux ; on y retrouve des anomalies squelettiques mais aussi
musculaires et des convulsions. Le bilan biologique révèle une
hypocalcémie, une hypophosphatémie, un taux de PTH élevéet une
forte phosphatase alcaline. Dans le VDDRII, les signes cliniques du
VDDRI sont retrouvés avec en plus une alopécie et un taux élevéde
1,25-dihydroxyvitamine D.
[40]
L’étude d’un cas présentant un
VDDRI montre des altérations dentaires avec, au niveau
radiologique, une pulpe dentaire large et des racines courtes et d’un
point de vue histologique, des altérations comparables àcelles
décrites précédemment dans le rachitisme hypophosphatémique
vitamine D-résistant. Cependant, il est intéressant de noter qu’il n’a
pas étéobservéd’abcès. Les signes dentaires du VDDRII sont
identiques àceux du VDDRI.
[40]
¶Hypophosphatasie
L’hypophosphatasie est une maladie héréditaire autosomale
récessive provoquant l’absence d’activitéphosphatase alcaline
(enzyme qui libère le phosphate en milieu basique). Il existe une
forme infantile de cette maladie oùles enfants décèdent avant
l’apparition des premières dents, une forme juvénile et une forme
adulte. Les malades présentent des déformations osseuses
ressemblant àcelles du rachitisme.
[11]
La dentine radiculaire est
particulièrement affectée. La dentine apicale est trèsfine avec des
fibres de collagène épaisses, des inclusions de débris cellulaires et
de nombreux espaces interglobulaires. Àla radiographie, la pulpe
dentaire de certains patients est extrêmement large, faisant penser à
des «dents en coquillage ».
[11]
¶Ostéogenèse imparfaite
L’ostéogenèse imparfaite est une maladie génétique oùla mutation
du gèneCOL1A1oudugène COL1A2 qui codent pour les chaînes
pro-a1etpro-a2 du collagène de type I provoque des altérations de
la minéralisation de cette protéine de la matrice osseuse et
dentinaire. Il existe quatre types d’ostéogenèse imparfaite classésen
fonction des signes cliniques, radiologiques et génétiques ou en
fonction du degréde mobilitédu malade. La dentinogenèse
imparfaite de type I est une forme de dysplasie dentinaire associée
àl’ostéogenèse imparfaite. Il convient de différencier la
dentinogenèse imparfaite de type I de la dentinogenèse imparfaite
de type II, anomalie génétique liéeàune mutation au niveau du
gène DSPP dont le phénotype est uniquement dentaire. Tous les
malades atteints d’ostéogenèse imparfaite ne présentent pas de
dentinogenèse imparfaite ; la prévalence de la dentinogenèse
imparfaite chez les patients atteints d’ostéogenèse imparfaite est de
8à40 %.
Qu’elle soit de type I ou de type II, les dents des patients atteints de
dentinogenèse imparfaite sont caractérisées par une couleur jaune
(en «sucre d’orge sucé») et un coefficient d’usure très rapide, l’émail
ayant disparu par attrition. Les dents temporaires sont plus atteintes
que les dents définitives. L’aspect radiologique est spécifique avec
des racines courtes, des couronnes bulbeuses dues àune constriction
cervicale et une oblitération de la chambre pulpaire. D’un point de
vue histologique, la dentine présente des zones amorphes
dépourvues de tubuli, ou des tubuli irréguliers et ramifiés, des
inclusions cellulaires et de la dentine interglobulaire. Cependant,
dans la dentinogenèse imparfaite de type I (associéeàl’ostéogenèse
imparfaite), ilyadetrès grandes variations en ce qui concerne la
gravitédes altérations de la dentine. Malmgren et Lindskog,
[46]
en
observant un grand nombre de patients atteints d’ostéogenèse
imparfaite, ont mis en évidence une corrélation entre la gravitéde
l’ostéogenèse imparfaite et la quantitéd’anomalies dentinaires. De
plus, cette étude montre qu’il y a peu de différences entre les dents
d’un même patient, que sur une même dent s’il y a une différence,
l’atteinte radiculaire est toujours plus accentuée que l’atteinte
coronaire, que la dentine circum-pulpaire est plus sévèrement
atteinte que la mantle dentine et que les patients qui présentent une
ostéogenèse imparfaite sans dentinogenèse imparfaite détectable
cliniquement ont des anomalies dentinaires plus importantes que
chez des patients sains.
Odontologie Facteurs étiologiques généraux de la pathologie pulpodentinaire 23-009-A-10
5
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%