La Biologie structurale

La Biologie structurale
La catalyse enzymatique covalente.
Peut-on prévoir la réactivité de l'acide aminé nucléophile impliqué ?
Guy Branlant
Summary
Résumé
La définition du codex existant entre structure primaire - structure 3D et l'efficacité
catalytique des enzymes opérant par catalyse covalente demeure un défi. En
particulier, il est difficile de prédire la réactivité des acides aminés nucléophiles dans
leurs environnements même quand la structure 3D est disponible. Tous les acides
aminés dotés de chaînes latérales qui montrent potentiellement des propriétés
nucléophiles sont capables de former un intermédiaire enzymatique covalent.
Généralement, ces enzymes sont catalytiquement efficaces. Etant donné la gamme
étendue de la nucléophilicité de ces acides aminés, cela implique un rôle majeur de
l'environnement protéique lors de la modulation de la réactivité chimique.
Dans la revue qui suit, les facteurs moléculaires impliqués dans l'augmentation, à
pH neutre, de la nucléophilicité de la cystéine essentielle de l'aldéhyde
déhydrogénase NAD(P)-dépendante sont caractérisés. Le changement de la cystéine
essentielle en acide aminé sérine conduit à une réduction drastique de l'efficacité
catalytique due à la faible nucléophilicité du groupement OH. D'un autre côte,
l'introduction d'une sélénocystéine conduit à un sélénoenzyme qui mime l'activité de
la sélénopéroxidase.
Ces résultats, ainsi que d'autres décrits dans la revue, montrent que : 1) des
solutions alternatives variées existent pour augmenter la nucléophilicité d'un résidu
cystéine à pH physiologique, 2) la structure cristalline de l'enzyme n'est pas
nécessairement représentative de sa forme biologiquement active, 3) l'analyse de la
structure 3D n'est pas suffisante pour prédire la réactivité d'un nucléophile, 4) une
étude biochimique est toujours nécessaire pour obtenir une meilleure
compréhension du caractère nucléophile, 5) la maîtrise du caractère nucléophile est
nécessaire mais pas suffisante pour créer de nouveaux biocatalyseurs efficaces
fonctionnant par un mécanisme covalent.
Introduction
Un des défis du décryptage du codex structure primaire-structure 3D-efficacité des
enzymes opérant par catalyse covalente est de prédire la réactivité des acides aminés
nucléophiles impliqués, dans le contexte de leur environnement protéique. Tous les
acides aminés à caractère nucléophile peuvent, en effet, former des intermédiaires

covalents. Généralement, les enzymes dans lesquelles ces nucléophiles sont impliqués,
présentent des efficacités catalytiques comparables qui contrastent avec le spectre de
réactivité chimique intrinsèque de ces nucléophiles à pH physiologique qui est très
variable, allant des résidus thréonine/sérine à faible réactivité à la sélénocystéine à très
forte réactivité.
Le processus limitant contrôlant l'efficacité enzymatique peut être associé a priori à
chacune des étapes du chemin réactionnel : l'attaque nucléophile sur le substrat incluant
la notion de positionnement efficace relatif du substrat et du nucléophile, la fixation et le
relargage des substrats, cosubstrats et produits, les changements conformationnels
éventuels associés, la stabilité et (ou) la réactivité différentes des nouveaux intermédiaires
réactionnels formés. Les efficacités relatives des différentes étapes sont façonnées au
cours de l'évolution pour atteindre un compromis conduisant à une efficacité globale
optimale. Ainsi, pour les enzymes à catalyse covalente impliquant des acides aminés à
faible réactivité, dont une étude cinétique approfondie est disponible, il apparaît que
l'étape limitante n'est jamais associée à un caractère nucléophile insuffisant du résidu
catalytique. Ceci implique donc que l'environnement protéique autour du résidu
catalytique augmente la réactivité du résidu, à pH physiologique.
Les enzymes à cystéine essentielle
Le pK apparent (pKapp) du thiol d'une cystéine intégrée dans un polypeptide non
structuré, en milieu aqueux, est environ de 8.7-9. Le fait que seule la forme thiolate soit
réactive implique que le pKapp du thiol soit abaissé de façon substantielle pour qu'une
enzyme à Cys soit catalytiquement efficace à pH neutre. Cette efficacité est aussi
fonction de l'accessibilité et de la nucléophilie du thiolate. Différents facteurs structuraux
et moléculaires peuvent conduire à modifier le pKapp d'un thiol. Ils incluent des effets
localisés à un acide aminé responsable par exemple d'une catalyse acide/base, d'une
interaction électrostatique spécifique ou des effets plus globaux, générés par un ensemble
de résidus d'une hélice ou de la chaîne principale peptidique ou d'un réseau de liaisons
hydrogène. A nouveau, ces effets, par delà leur influence sur le pKapp, peuvent aussi
fortement moduler la réactivité du thiolate. Une interaction entre un résidu chargé
positivement et un thiolate doit conduire à une diminution de la nucléophilie du thiolate.
A l'inverse, la présence d'un résidu chargé négativement dans l'environnement proche du
thiolate devrait amener à augmenter sa réactivité. Il s'avère donc que, pour appréhender la
réactivité d'un thiolate, il est nécessaire de connaître non seulement son pKapp, mais
aussi sa nucléophilie et son accessibilité, facteurs essentiels pour évaluer l'efficacité de la
formation de la liaison entre le thiolate et le substrat. C'est dans ce contexte que se situe
le projet de notre groupe de recherche sur les aldéhydes déshydrogénases à cofacteur
NAD(P), enzyme dont le mécanisme implique une catalyse covalente via une cystéine
essentielle.
Présentation des aldéhydes déshydrogénases à cofacteur NAD(P)
1. Préambule
Les composés aldéhydiques sont, de part la présence de leur fonction aldéhyde, des
entités chimiquement réactives et donc potentiellement toxiques. In vivo, ils dérivent du
processus de biotransformation de métabolites d'origine endogène ou exogène avec un
large spectre de structures différentes. De source endogène, ils englobent les aldéhydes

provenant du métabolisme d'acides aminés, d'amines, de polyoses, de stéroïdes ou de
lipides. De source exogène, ils dérivent de la biotransformation de xénobiotiques ou de
vitamines. Plusieurs classes d'enzymes d'oxydoréduction sont impliquées dans leur
métabolisme, c'est en particulier le cas des aldéhydes déshydrogénases (ALDH) à
cofacteur NAD(P) qui catalysent l'oxydation des aldéhydes en acides activés
phosphorylés ou non activés.
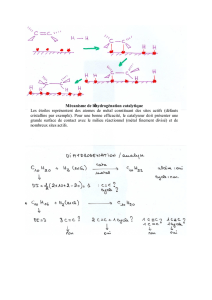
2. Présentation du mécanisme catalytique
Les ALDH présentent un mécanisme de catalyse chimique à deux étapes, une étape
d'oxydoréduction conduisant à la formation d'un intermédiaire thioacylenzyme suivie
d'une étape soit de phosphorolyse [ce sont les ALDH phosphorylantes dont fait partie la
glycéraldéhyde-3-phosphate déshydrogénase glycolytique (GAPDH)], soit d'hydrolyse
(ce sont les ALDH non phosphorylantes) (figure 1). La première étape commune aux
deux types d'ALDH se fait via la formation d'un intermédiaire thiohémicétal avec une
Cys essentielle puis transfert d'hydrure sur le noyau nicotinamidium du cofacteur pour
former l'intermédiaire thioacylenzyme. La bonne efficacité enzymatique des ALDH, à pH
physiologique, implique donc :
a) une bonne nucléophilie de la cystéine essentielle à pH neutre, donc un environnement
protéique adapté pour révéler cette réactivité ;
b) un transfert efficace de l'hydrure de l'intermédiaire thiohémicétal vers le pyridinium,
donc la participation d'un acide aminé jouant le rôle de base, à moins que l'environnement
protéique soit de type oxyanion et tel qu'il abaisse fortement le pKapp de l'intermédiaire,
suffisamment pour ne pas nécessiter l'intervention d'une catalyse basique ;
c) la stabilisation des états de transition et des intermédiaires formés ;
d) la présence pour les ALDH phosphorylantes d'un site de fixation du phosphate pour
permettre une phosphorolyse efficace et pour les ALDH non phosphorylantes, d'un site
adapté pour activer la nucléophilie de la molécule d'eau.
3) Caractéristiques de la cystéine essentielle des GAPDH d'origine bactérienne
3. 1. Comment la réactivité de la Cys essentielle est-elle augmentée à pH neutre ?
L'étude faite sur la GAPDH d'Escherichia coli sous forme apoenzyme (en absence de
cofacteur NAD) montre que la cystéine essentielle 149 est impliquée dans une interaction
de type paire d'ions avec l'His176 essentielle du site actif avec un pKapp de 5.7 pour la
Cys149 et de 7.9 pour l'His176 (figure 2 ). Ceci a été démontré sur la base d'études
spectroscopiques qui montrent :
a) pour le type sauvage, une courbe biphasique de l'absorbance de l'ion thiolate en
fonction du pH, décomposable en 2 monoexponentielles avec des pKapp de 5.7 et 7.9 ;
b) pour le mutant His176Asn, une courbe monophasique avec un pKapp de 7.1.
L'utilisation de sondes cinétiques pour titrer les thiolates a non seulement permis de
confirmer les résultats précédents (pKapp de 5.5 pour la Cys149 et de 8.25 pour l'His176
dans le type sauvage et pKapp de 7.4 pour la Cys149 dans le mutant His176Asn) mais a
aussi montré que le pouvoir nucléophile, qui mesure la réactivité de la Cys149 dans la
paire d'ions, est suffisant pour que l'attaque nucléophile ne soit pas limitante dans le
processus catalytique à pH neutre, même si le pouvoir nucléophile est diminué d'un
facteur 20 par rapport à celui d'un thiolate " libre " (1).

Des études faites sur la GAPDH de Bacillus stearothermophilus donnent des résultats
similaires avec un pKapp de la Cys149 de 5.5 dans le type sauvage et de 8 dans le mutant
His176Asn. L'addition du cofacteur favorise la formation d'une paire d'ions plus efficace
avec un pKapp de la Cys149 de 4.45. Un tel résultat montre que la fixation du cofacteur
doit induire un changement conformationnel, autour de la Cys149, rapprochant l'His176
de la Cys149, augmentant ainsi la stabilité de la paire d'ions.
3.2. Ces résultats étaient-ils prévisibles à partir de l'inspection des structures RX
actuellement disponibles ?
Si, dans le voisinage de la Cys149, on trouve effectivement l'His176 dont le N pointe
vers le groupement thiol, il n'était cependant pas possible de prédire que cette interaction
conduirait à la formation d'une paire d'ions aussi efficace. Les distances entre la Cys149
et l'His176 déduites des modèles de structure 3D - 3.6 Å dans l'holo structure - (2, 3) sont
en effet incompatibles avec la formation d'une paire d'ions efficace comme cela a été
démontré par les études biochimiques. Ces résultats suggèrent donc que la structure
déterminée par RX n'est pas tout à fait représentative de celle qui est biologiquement
efficace. Des études récentes couplant une approche hybride de calcul de mécanique
quantique/dynamique moléculaire confirment cette hypothèse en montrant que la distance
Cys149 - His176 est en fait de 2.9 Å soit 0.7 Å plus courte que celle déduite des
structures 3D (4).
Si l'His176 joue donc un rôle essentiel dans la révélation de la nucléophilie de la Cys149,
d'autres facteurs structuraux doivent intervenir pour abaisser le pKapp de la Cys149. En
effet, le pKapp de la Cys149 dans le mutant His176Asn est de 1.4 (E. coli) et 0.7 (B.
stearothermophilus) unités plus bas que celui observé dans le glutathion. Les différences
observées de 1.4 et de 0.7 doivent refléter des différences subtiles entre les structures des
deux sites des GAPDH au voisinage de la Cys149, différences qui sont impossibles à
prévoir par la seule inspection des deux structures (2, 3). Néanmoins, l'inspection des
structures 3D des GAPDH montre que la Cys149 se trouve aussi à l'extrémité N
terminale d'une hélice . Ceci suggérait donc une contribution du dipôle généré par
l'hélice alpha pour abaisser le pKapp de la Cys149 comme cela avait été montré
récemment sur des modèles chimiques (5). L'obtention par ingénierie d'un fragment
protéique de la GAPDH de B. stearothermophilus ne contenant que le domaine du
cofacteur et une partie de l'hélice alpha sous-tendant la Cys149, mais ne possédant pas le
domaine catalytique et donc l'His176, a permis de titrer un pKapp de 8.3 pour la Cys149.
La contribution de l'hélice alpha dans l'abaissement du pKa de la Cys149 serait donc
d'environ 0.4 unité, valeur différente de celle estimée à partir de modèle chimique (1.4
unités), résultat qui, à nouveau sur la base de la structure 3D, était difficile à prédire.
3.3. Le mécanisme d'activation de la Cys 149 dans les GAPDH bactériennes peut-il être
étendu à toutes les GAPDH ?
Les résultats précédents ont montré qu'un des facteurs essentiels impliqué dans
l'activation de la Cys 149 était une His. L'isolement de GAPDH d'archaea,
phylogénétiquement distante des GAPDH bactériennes et eucaryotes, a permis
d'entreprendre une étude comparative de l'évolution des sites actifs des deux types de
GAPDH. La présence d'une His proche de la Cys 149 a pu être montrée sans que la
formation d'une paire d'ions n'ait pu être mise en évidence dans l'apoenzyme. Mais le
résultat le plus intéressant de cette étude est que cette His se trouve positionnée ailleurs
dans la structure primaire (6). Un tel résultat suggère une évolution convergente des deux

sites actifs, l'activation de la Cys 149 ainsi que le mécanisme de transfert d'hydrure
nécessitant la présence d'une His proche spatialement.
3.4. La présence d'une cystéine 149 très réactive est-elle suffisante pour générer une
enzyme catalytiquement efficace ?
En présence de son substrat biologique, le glycéraldéhyde-3-phosphate (G3P), la GAPDH
bactérienne est très efficace, l'étape limitante n'étant pas associée à l'attaque nucléophile.
L'utilisation de substrat comme l'érythrose-4-phosphate (E4P) (1 groupement CHOH de
plus que le G3P) conduit à une très faible activité due à un site de spécificité structurale
non adapté, résultat en soi qui n'a rien de surprenant. Par contre, une enzyme GAPDH
" like " d'E. coli ayant les mêmes acides aminés essentiels pour la catalyse montre une
forte efficacité enzymatique avec l'E4P et une faible efficacité avec le G3P (7). Nos
études cinétiques ont prouvé que la faible efficacité en présence de G3P était due à la
formation d'un complexe ternaire enzyme-NAD-G3P peu efficace pour le transfert
d'hydrure, la nucléophilie de la Cys149 n'étant pas modifiée. Ceci suggère que quelques
mutations, vraisemblablement dans le site de spécificité structurale, changent
drastiquement l'efficacité enzymatique, et ceci sans changer le pouvoir nucléophile de la
Cys essentielle (la nature et le rôle de ces mutations n'ont pu être prédits de la structure
3D simulée à partir de celle de la GAPDH d'E. coli). De tels résultats montrent que la
conception d'enzymes efficaces évoluant par catalyse covalente implique non seulement
de maîtriser le pouvoir nucléophile de l'acide aminé essentiel qui doit être suffisant, mais
aussi d'adapter le site de spécificité structurale à la structure du substrat que l'on veut
utiliser.
4. Que se passe-t-il si on introduit une sérine à la place de la Cys149 dans les
GAPDH bactériennes ?
Intrinsèquement, une sérine est moins réactive qu'une cystéine. Cependant, les protéases
à sérine montrent des efficacité catalytiques élevées, ce qui implique que le caractère
nucléophile de la sérine est fortement augmenté par son environnement protéique. La
présence d'une diade de diade Ser/His et His/Asp est décrit comme étant l'élément moteur
de cette activation. Généralement, changer une cystéine essentielle en sérine ou
inversement une sérine essentielle en cystéine conduit à une forte perte d'efficacité
enzymatique. A part un caractère nucléophile insuffisant, d'autres raisons peuvent être
avancées pour expliquer la perte d'activité : un positionnement non adéquat du substrat
vis-à-vis du nucléophile (distance non optimale), une stabilité et une réactivité différentes
des intermédiaires réactionnels formés.
Jusqu'à présent, aucune étude approfondie de l'influence d'un site actif sur la nucléophilie
d'un résidu sérine introduit à la place d'une cystéine essentielle n'avait été entreprise. Nos
résultats montrent clairement que le site de la GAPDH n'est pas adapté pour augmenter la
nucléophilie d'une Ser bien que l'His176 soit proche spatialement, histidine qui aurait pu
favoriser cette nucléophilie comme dans le cas des protéases à Ser. En fait, l'activité
catalytique est diminuée d'un facteur104 (8). Ce résultat n'est cependant pas forcément
extrapolable. En effet, si introduire une Cys à la place d'une Ser dans une protéase
conduit à une forte diminution de l'activité enzymatique, cette diminution n'est pas liée à
un faible pouvoir nucléophile de la Cys introduite. Il a été montré que la Cys possédait
une forte réactivité grâce à la formation d'une paire d'ions avec l'His essentielle, comme
dans la papaïne (9). En d'autres termes, le site actif des protéases à Ser est adapté pour
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%