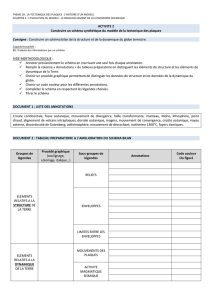
Outil d`aide au clonage positionnel Bioinformatic tool for positionnal

Outil d’aide au clonage positionnelG. Ricard, S. Gallina et P. FroguelJOBIM 2002
Outil d’aide au clonage positionnel
Bioinformatic tool for positionnal cloning
Guénola Ricard†* Sophie Gallina† Philippe Froguel†
† Institut de Biologie de Lille, CNRS UPRES A 8090, 1, rue du Pr. Calmette, 59000 Lille.
* DESS EGOISt (Etude des Génomes : Outils Informatiques et Statistiques)– Université de Mont Saint Aignan
Courriel : {guenola.ricard, sophie.gallina, philippe.froguel}@mail-good.pasteur-lille.fr
Résumé
Nous avons développé un outil bio-informatique pour inventorier les SNP d'une région d'intérêt, et les classer en
fonction des annotations associées à leur position (gène connu ou prédit, exon, région régulatrice ou intron,
transcrit, séquence homologue dans une autre espèce…). Nous avons développé cet outil dans la mesure où les
serveurs d'annotations existants (Golden Path, Ensembl, NCBI…) ne permettent pas de rechercher des
"combinaisons d'annotations", par exemple «SNP se trouvant dans un exon». Cet outil est conçu pour prendre
en compte l'évolution des versions de l'assemblage du génome humain et des annotations qui y sont associées. Il
permettra aux biologistes d’obtenir soit un ensemble complet de résultats, soit un état des mises à jours
(modification de position, ajout d'annotations …). Afin de favoriser l'intégration de différentes sources
d'annotations, les résultats produits seront exportés selon le protocole DAS (Distributed Annotation System).
Mots-clés : classification des SNP, visualisation des annotations, DAS (système d'annotations distribuées).
Abstract
We developed a bio-informatic tool to explore SNPs in a region of interest, and classify these SNPs according to
their position in other annotations (known gene, prediction, exon, intron, mRNA,..) We developed this tool
because existing annotation servers (Golden Path, Ensembl, NCBI …) do not allow user to search for "combined
annotation" (ie “SNP in exon”). This tool will manage regular updates from new versions of the human draft
and associated annotations and will supply biologists with a complete list of results or reports of modifications
in positions, additional annotations…. In order to promote annotations integration, results will be exported
using DAS protocol (Distributed Annotation System).
Keywords: SNPs classification, annotations visualisation, DAS (Distributed Annotation System).
1 Introduction
Le principal axe de recherche de l’UPRES A 8090 de l’Institut de Biologie de Lille est l’étude
génétique des maladies multifactorielles telles que le diabète de type 2 et l’obésité. Pour trouver les
gènes impliqués dans ces maladies, les chercheurs peuvent utiliser 2 stratégies :
1. Gène candidat : Tester l'implication d'un gène déjà connu et ayant un rapport avec la pathologie
2. Génétique inverse :
• Genome Scan : Tester l'ensemble du génome par des méthodes d'analyse de liaison pour
localiser des régions chromosomiques liées à la maladie, c'est à dire susceptibles de contenir
les gènes de prédisposition à la maladie.
• Clonage positionnel : Rechercher les gènes connus ou inconnus se trouvant dans cette région
et tester leur éventuelle association avec la maladie étudiée par des analyses de déséquilibre de
liaison avec des polymorphismes de type SNP (Single Nucleotide Polymorphisms), qui sont
très fréquents dans le génome.
L'outil que nous avons développé a pour but de faciliter l'étape de clonage positionnel en automatisant
la sélection de SNP intéressants dans une région.
JOBIM 2002 325

2 Problématiques
Pour rechercher les gènes et les annotations localisés dans une région, les chercheurs utilisent
différents serveurs d’ annotations tels que le Genome Browser de l’UCSC [1] , Ensembl [2], Map
viewer [3] du NCBI, ...
Durant cette recherche, plusieurs problématiques se dégagent :
1. Les annotations sont distribuées sur plusieurs serveurs. La phase d’ étude in silico de la région est
donc longue et fastidieuse : on se heurte à un grand nombre d’annotations redondantes.
2. De plus ces données évoluent au fur et à mesure de l’apparition de nouvelles versions de
l’ assemblage du génome. Il est important que les chercheurs puissent prendre connaissance de ces
changements d’une version à l’ autre, sans avoir à refaire entièrement l’ organisation de leur région.
3. Pour détecter des associations préférentielles entre un allèle et un gène impliqué dans la maladie,
nous utilisons des SNP. Il existe environ un SNP tous les 800 paires de bases. Pour une région de
5Mb, on trouve donc dans les bases publiques plus de 6000 SNP, dont un grand nombre «non
confirmés». Typer ces 6000 SNP sur la population étudiée (environ 200 individus) est très coûteux
et inutile. L'alternative consiste à ne typer que les SNP les plus intéressants, sélectionnés et classés
en fonction de l’ environnement dans lesquels ils se trouvent (par exemple, exon d'un gène).
L'utilisation d'outils standards de visualisation avec plusieurs types d'annotations (SNP, gènes
connus, prédictions, mRNA) est suffisante pour effectuer ce travail de sélection sur des petites
portions (< 1Mb), mais s'avère inexploitable au delà.
4. De plus au fur et à mesure de l'avancement du clonage positionnel, nous disposons des résultats
obtenus sur les premiers SNP analysés. Nous souhaitons pouvoir intégrer ces résultats, ou toute
autre donnée locale pertinente, avec la sélection de SNP obtenus par cet outil.
3 Réalisation
Ces problématiques bien qu’ apparemment distinctes, ne peuvent être traitées séparément. C’ est à la
suite de cette constatation que nous avons développé une base de données relationnelle et des
procédures pour traiter et intégrer les différentes annotations que nous souhaitons associer aux SNP.
Après avoir rapatrié les données d’ annotations depuis le serveur de l'UCSC, nous avons effectué un
traitement pour attacher à un SNP, toutes les informations qui lui sont relatives, puis nous avons créé
une interface web conviviale permettant une interrogation intuitive de cette base.
Les biologistes peuvent ainsi interroger la base contenant les données associées à leur région en
sélectionnant également les SNP qu’ils souhaitent voir afficher. Les possibilités de sélection sont :
• Une sous-région
• La localisation des SNP que l’ on souhaite afficher :
1. Dans les gènes (mais également de façon plus spécifique dans les exons, régions flanquantes
et régulatrices, introns)
2. Dans les séquences d’ ARNm et les EST
3. Dans les prédictions (mais également de façon plus spécifique dans les exons prédits, régions
flanquantes et régulatrices prédites, introns prédits)
L’ utilisateur peut croiser les requêtes et demander par exemple à afficher les SNP contenus dans un
gène prédit par au moins deux logiciels de prédiction et retrouvé dans un mRNA.
Il est alors possible d’ afficher un grand nombre d’ informations associées aux annotations (dont le
nucléotide changé et éventuellement l’ acide aminé modifié) et des liens web vers les bases de données
publiques.
Comme évoqué précédemment, les biologistes obtiennent une grande quantité de données qui sont
parfois mises à jour sur les serveurs d’ annotations. Notre objectif est d’éviter de leur fournir à nouveau
une grande liste de données. Nous réalisons par conséquent une nouvelle base que nous comparons
avec l’ancienne afin de pouvoir leur donner uniquement les modifications.
G. Ricard, S. Gallina et P. Froguel
JOBIM 2002326

4 Résultats
Rappel des parameters selectionnés
intervalle 23533946 - 23543986
paramètres typeSNP : all,
Nombre de réponses :
18
Nom
du
SNP
Position
du SNP Distance Localisation Origine Commentaire Variation Validation Prédiction Genscan Prédiction Ensembl Prédiction Fgenesh
915861 23533946 0 AF086441 mRNA Exon 1 (23533901-
23534351) G/T confirmé Intronique NT_011512.521
2298368 23534544 598 NT_011512.521 Prédiction G/A Intronique NT_011512.521
2298369 23535095 551 NT_011512.521 Prédiction C/G Intronique NT_011512.521
2282471 23535562 467 NT_011512.521 Prédiction C/T Intronique NT_011512.521
2282472 23535707 145 NT_011512.521 Prédiction A/G Intronique NT_011512.521
2282473 23536007 300 NT_011512.521 Prédiction A/T Intronique NT_011512.521
2829806 23539003 2996 NM_017446
(PRED22) gène connu Intronique T/G Intronique ENST00000284967 Intronique mais proche de
l’exon 3 (23538827-
23538916) C21000071
2829807 23539900 897 NM_017446
(PRED22) gène connu Intronique mais proche
de l’exon 2 (23539961-
23540009) T/C Intronique mais proche de
l’exon 2 (23539961-23540009)
ENST00000284967
Intronique mais proche de
l’exon 4 (23539961-
23540009) C21000071
1539764 23543403 3503 NM_017446
(PRED22) gène connu Intronique C/T Intronique NT_011512.522 Intronique ENST00000284967 Intronique C21000071
…
1539765 23543797 28 NM_017446
(PRED22) gène connu Intronique A/G Intronique NT_011512.522 Intronique ENST00000284967 Intronique C21000071
2248298 23543885 88 NM_017446
(PRED22) gène connu Intronique mais proche
de l’exon 3 (23543938-
23544092) G/A Intronique mais proche de
l’exon 2 (23543938-23544092)
NT_011512.522
Intronique mais proche de
l’exon 3 (23543938-23544092)
ENST00000284967
Intronique mais proche de
l’exon 5 (23543938-
23544092) C21000071
2829809 23543914 29 NM_017446
(PRED22) gène connu Intronique mais proche
de l’exon 3 (23543938-
23544092) C/A Intronique mais proche de
l’exon 2 (23543938-23544092)
NT_011512.522
Intronique mais proche de
l’exon 3 (23543938-23544092)
ENST00000284967
Intronique mais proche de
l’exon 5 (23543938-
23544092) C21000071
1135638 23543962 48 NM_017446
(PRED22) gène connu Exon 3 (23543938-
23544092) A/G +++ exon 2 (23543938-
23544092) NT_011512.522 +++ exon 3 (23543938-23544092)
ENST00000284967 +++ exon 5 (23543938-
23544092) C21000071
10576 23543986 24 NM_017446
(PRED22) gène connu Exon 3 (23543938-
23544092) T/C +++ exon 2 (23543938-
23544092) NT_011512.522 +++ exon 3 (23543938-23544092)
ENST00000284967 +++ exon 5 (23543938-
23544092) C21000071
FIG. 1 – Consultation des SNP sélectionnés
La figure 1 montre un extrait des résultats obtenus lors d'une interrogation sur les SNP localisés dans
une région du chromosome 21. La seule restriction entrée en paramètre a été l’ intervalle 23 533 946 –
23 543986 ; l’utilisateur a demandé à voir tous les SNP de cette région avec leur localisation relative
aux autres annotations. La couleur rouge est utilisée pour faire ressortir les SNP se trouvant dans les
exons des gènes connus, la couleur verte, pour faire ressortir les SNP se trouvant sur le mRNA. Le
Nom du SNP comporte un lien vers le site SNPper [4] qui est un site dédié aux SNP. La colonne
Position du SNP donne la position par rapport à l'assemblage du NCBI. La colonne Origine spécifie
s'il s'agit d'un gène connu, prédit ou identifié à partir d'une région homologue dans une autre espèce.
La colonne Commentaire donne des précisions sur la localisation du SNP (partie codante ou non de
l’ annotation). La colonne Variation précise le changement de nucléotides et éventuellement celui de
l’ acide aminé si l’ information est disponible. La colonne Validation précise si le SNP est confirmé,
suivant les données de la base dbSNP [5]. Les 3 colonnes suivantes concernent les données fournies
par les logiciels de prédiction Genscan [6], Ensembl et Fgenesh [7].
Des critères plus stringents auraient pu être spécifiés dans le formulaire, tels que « SNP se trouvant
dans les mRNA et prédits par au moins 2 logiciels de prédiction, parmi Genscan, Ensembl et
Fgenesh » ou bien « SNP se trouvant dans les exons ou les parties flanquantes des gènes connus » et
ceci pour le chromosome 21 entier ou seulement une partie de ce chromosome.
Nous avons choisi une stratégie permettant une réponse instantanée pour l’ utilisateur, ce qui nécessite
la création d’ une table de pré-traitement contenant toutes les informations relatives à chaque SNP. Les
temps d’ exécution des différentes étapes de construction de cette table dépendent de la taille de la
région et du nombre d’ annotations. A titre d’ exemple le traitement de la totalité du chromosome 21
Outil d’aide au clonage positionnel
JOBIM 2002 327

(44 Mb) prend environ 1 heure. Les algorithmes de traitement sont en cours d’optimisation. La place
mémoire nécessaire sur le disque est de l’ ordre de 4 Mo pour le chromosome 21 (34061 SNP).
5 Intégration de l'outil dans un ensemble de développement
L'objectif est que les biologistes puissent visualiser sur leur région, en même temps les données issues
des serveurs publiques, les SNP sélectionnés avec leur niveau d'intérêt, et les résultats des SNP déjà
analysés dans le laboratoire. En d'autres termes, il s'agit d'intégrer des annotations publiques et locales,
provenant de différents serveurs. Le protocole DAS [8] (Distributed Annotation System) répond à
cette préoccupation, en permettant l’ intégration à la volée, c'est à dire au niveau client, de plusieurs
sources de données, sans nécessité d'interaction entre les serveurs concernés. Ce protocole est basé sur
le principe d'un serveur de référence unique pour les données d'assemblage, et de multiples serveurs
d'annotations. C'est pourquoi nous avons choisi ce protocole pour intégrer nos outils.
L'outil de classification des SNP utilisera ce protocole pour exporter ses résultats, c'est à dire la liste
des SNP sélectionnés et les diverses informations associées. Les SNP sélectionnés seront donc vus
comme un type d'annotation supplémentaire.
Par ailleurs les résultats des analyses de différents polymorphismes (micro-satellites, SNP) sont
également interrogeable via un serveur d'annotations. Actuellement, ces résultats relatifs aux
phénotypes sont stockés dans une base de données relationnelle, qui comporte des informations sur les
populations utilisées, les phénotypes étudiés, les localisations des régions ou des gènes, les méthodes
utilisées, les scores obtenus et la source de ces résultats (laboratoire ayant produit ces résultats,
publication). Les résultats disponibles sur d'autres espèces (souris, rat) y seront bientôt intégrés. Cette
base pourra exister en 2 versions : une version de travail locale au laboratoire, et une version publique
accessible par Internet pour les résultats publiés.
Ces 2 types de résultats locaux étant accessibles par un serveur DAS, tout client DAS pourra les
visualiser conjointement aux annotations publiques.
6 Implémentation
Ce projet a été développé sur un serveur SUN Enterprise 3500 sous Solaris. Le SGBD utilisé est
MySQL. Les procédures d'importation des données à partir des bases publiques sont implémentées en
Perl avec les modules DBI, LWP. Les procédures d'intégration entre les différentes annotations sont
développées en PHP. L'interface utilisateur pour l'accès aux résultats est développée en PHP. Enfin,
nous utilisons le logiciel LDAS [9] comme serveur DAS.
Références
[1] Golden Path http://genome.ucsc.edu,http://www.cse.ucsc.edu/~kent/
[2] Ensembl http://www.ensembl.org/Homo_sapiens
[3] Mapviewer http://www.ncbi.nlm.nih.gov/cgi-bin/Entrez/hum_srch
[4] SNPper http://bio.chip.org:8080/bio
[5] dbSNP http://www.ncbi.nlm.nih.gov/SNP/
[6] Genscan http://genes.mit.edu/GENSCANinfo.html
[7] Fgenesh http://genomic.sanger.ac.uk/gf/Help/fgenesh.html
[8] LINCOLN STEIN, SEAN EDDY, ROBIN DOWELL, Distributed Sequence Annotation System (DAS)
http://stein.cshl.org/das july 26, 2000.
[9] LDAS http://biodas.org/servers
G. Ricard, S. Gallina et P. Froguel
JOBIM 2002328
1
/
4
100%