revue - John Libbey Eurotext

Journal Identification = VIR Article Identification = 0462 Date: October 19, 2012 Time: 1:45 pm
revue
Virologie 2012, 16 (5) :315-29
Cytomégalovirus humain et cancers
Quentin Lepiller1,2,3
Samira Fafi-Kremer1,2,3
Franc¸oise Stoll-Keller1,2,3
Georges Herbein4,5,6
1Hôpitaux universitaires de Strasbourg,
laboratoire de virologie, 3, rue Koeberlé,
67000 Strasbourg, France
2Inserm, U748, 67000 Strasbourg,
France
3Université de Strasbourg, 67000
Strasbourg, France
4CHU de Besanc¸on, laboratoire de
virologie, 25030 Besanc¸on, France
5Université de Franche-Comté,
EA 4266 Agents pathogènes et
inflammation, 25030 Besanc¸on, France
6SFR FED 4234, 25030 Besanc¸on,
France
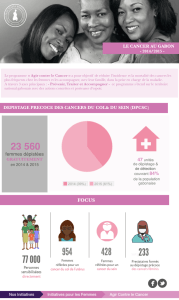
Résumé. Le cytomégalovirus humain (HCMV) est un virus aux multiples
facettes dont la pathogénicité ne cesse d’être réévaluée. Si son implication dans
la carcinogenèse est restée longtemps controversée, l’amélioration récente de la
sensibilité des techniques de détection a permis de le mettre en évidence dans
les tissus tumoraux de plusieurs cancers. Son influence, en tant qu’initiateur
ou promoteur de l’oncogenèse, mérite d’être envisagée. Le HCMV favorise
l’inflammation, pilier de la carcinogenèse, ainsi que l’échappement immunitaire.
En infectant la cellule tumorale et/ou les cellules du microenvironnement tumoral
et en modulant le cycle cellulaire, la survie, l’apoptose, le métabolisme cellulaire,
l’angiogenèse, l’adhésion et la migration cellulaire, le HCMV pourrait fournir à
la tumeur en formation des armes supplémentaires pour promouvoir son déve-
loppement. Les preuves d’un tel effet oncomodulateur du HCMV n’ont cessé de
s’accumuler depuis plusieurs années. Parallèlement, certains arguments conti-
nuent de plaider en faveur d’un rôle direct du HCMV sur l’initiation des tumeurs.
Qu’il soit initiateur ou promoteur, le rôle potentiel du HCMV dans la carcino-
genèse permet d’envisager de nouvelles cibles thérapeutiques dans le traitement
des cancers et offre un regard nouveau sur les relations complexes entre virus et
cancers.
Mots clés : herpesvirus, carcinogenèse, inflammation, échappement immunitaire
Abstract. Human cytomegalovirus (HCMV) has been increasingly involved in
carcinogenesis over the last decade. HCMV is present on the tumor site in a
large proportion of several cancers and enhances both inflammation and immune
escape. By acting on the tumor cells and/or the tumor microenvironment, HCMV
may behave like an “oncomodulator” to influence cell cycle progression, survival,
apoptosis, angiogenesis, cellular metabolism and tumor invasivity. Beside this
suspected role in tumor promotion, several data continue to argue for a direct
role of HCMV in tumor initiation. Whether HCMV is initiator or promoter,
its potential involvement in carcinogenesis may provide new therapeutic targets
in cancer treatment and offers a new perspective on the complex relationship
between viruses and cancers.
Key words: herpeviruses, carcinogenesis, inflammation, immune escape
Introduction
La relation entre virus et cancers est une histoire complexe
qui débuta il y a tout juste un siècle lorsque Peyton Rous
démontra que des extraits tumoraux filtrés acellulaires pré-
parés à partir de sarcomes de poulets pouvaient reproduire
la tumeur d’origine lorsqu’ils étaient injectés à un animal
Tirés à part : Q. Lepiller
sain. Ces filtrats acellulaires réalisés par Rous contenaient
un virus, le virus du sarcome de Rous (RSV), qui devint
ainsi le premier virus oncogène découvert et qui valut à
Peyton Rous de recevoir le prix Nobel en 1966. Depuis,
le rôle oncogène de nombreux virus à ARN ou à ADN
a été largement démontré et a permis d’envisager des
cibles vaccinales et thérapeutiques efficaces dans certains
cancers comme le cancer du col de l’utérus (impliquant
des papillomavirus humains) ou l’hépatocarcinome (pou-
vant résulter d’une infection par les virus de l’hépatite B
doi:10.1684/vir.2012.0462
Virologie, Vol 16, n◦5, septembre-octobre 2012 315
Pour citer cet article : Lepiller Q, Fafi-Kremer S, Stoll-Keller F, Herbein G. Cytomégalovirus humain et cancers. Virologie 2012; 16(5) : 315-29 doi:10.1684/vir.2012.0462
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

Journal Identification = VIR Article Identification = 0462 Date: October 19, 2012 Time: 1:45 pm
revue
et de l’hépatite C) [1]. Les virus peuvent agir en tant
qu’initiateurs des cancers ou en tant que promoteurs, notam-
ment en induisant une inflammation chronique favorisant
le passage de cellules prénéoplasiques vers le développe-
ment de tumeurs invasives. Il est généralement admis que
plus de 20 % des cancers seraient ainsi sous-tendus par un
processus inflammatoire chronique [2]. À l’échelle de la
population mondiale, il a été récemment estimé que deux
millions de nouveaux cas de cancers (sur un nombre total de
12,7 millions) étaient directement imputables à des agents
infectieux au cours de l’année 2008 [3]. De nombreux pro-
grès restent cependant à faire pour identifier l’ensemble des
virus impliqués dans la survenue de cancers et comprendre
les mécanismes moléculaires liant directement ou indirec-
tement les infections virales et la carcinogenèse.
À la différence d’autres herpesvirus comme le virus
d’Epstein-Barr (EBV) ou l’herpesvirus humain 8 (HHV8),
le cytomégalovirus humain (HCMV) n’est souvent pas
considéré comme un virus oncogène. Le HCMV est un
-herpesvirus aux multiples facettes dont la pathogénicité
ne cesse d’être réévaluée. En effet, en dehors du contexte
de primo-infection au cours de la grossesse, le HCMV
fut considéré pendant des décennies comme un virus peu
pathogène chez l’hôte immunocompétent, responsable le
plus souvent d’une primo-infection asymptomatique, par-
fois responsable de syndromes mononucléosiques [4]. Avec
l’accroissement des situations d’immunosuppression, liées
au développement des transplantations de moelle osseuse
et d’organes solides, à l’usage de traitements immunosup-
presseurs et à l’émergence du sida, le pouvoir pathogène
du HCMV apparut sous un jour nouveau. Chez les patients
immunodéprimés, le HCMV est impliqué à la fois dans
des complications infectieuses aiguës telles que des pneu-
monies, des œsophagites, des colites ou des rétinites, mais
également dans des complications à long terme comme des
complications vasculaires, des pathologies auto-immunes,
des maladies du greffon contre l’hôte et des rejets de greffe
[4, 5]. L’infection endothéliale par le HCMV est impli-
quée, via la libération de médiateurs de l’inflammation et
de facteurs de croissance, dans le développement accé-
léré de pathologies vasculaires comme l’athérosclérose,
associé à une inflammation chronique avec prolifération
des cellules musculaires lisses [6]. Par analogie avec les
complications tardives observées chez les sujets immuno-
déprimés, le HCMV est suspecté de participer à des pro-
cessus inflammatoires similaires chez les sujets immuno-
compétents dont l’athérosclérose, certaines maladies
inflammatoires chroniques (comme la maladie de Crohn),
ou encore des cancers [5]. Les arguments en faveur de
l’implication du HCMV dans la promotion des cancers
n’ont cessé de s’accumuler, offrant un regard nouveau
sur la complexité des relations entre virus et cancers
[5, 7, 8].
Nous nous proposons de présenter successivement les
arguments en faveur d’une participation du HCMV aux pro-
cessus tumoraux. Nous comparerons également brièvement
le HCMV à un autre herpesvirus dont le pouvoir oncogène
est bien établi, l’EBV, avant de nous interroger sur la place
du HCMV comme cible thérapeutique dans le traitement
des cancers.
Le cytomégalovirus humain
est présent au cœur
de l’inflammation et de la tumeur
Après une phase de primo-infection, le HCMV établit chez
son hôte une phase de latence. Chez le sujet immunocom-
pétent, cette phase de latence est ponctuée de réactivations
régulières efficacement contrôlées par le système immuni-
taire [4]. Les monocytes et certaines cellules CD34+de la
lignée myéloïde constituent un site majeur de latence du
HCMV [9]. La réplication virale y est abortive ou limitée
à l’expression de certains gènes. En revanche, l’activation
des monocytes et leur différenciation en macrophages,
s’accompagnent d’une reprise du cycle de réplication virale
[10]. La capacité du virus à se répliquer est donc étroitement
liée à l’état de différenciation de la cellule infectée. Certains
auteurs suggèrent que le HCMV serait véhiculé jusqu’au
cœur du processus inflammatoire tumoral par les monocytes
puis réactivé lors de la différenciation des monocytes en
macrophages. Le HCMV infecterait alors les cellules per-
missives environnantes (dans le cas des cancers : cellules
tumorales ou prétumorales, cellules stromales et cellules
immunes) et agirait comme cofacteur dans l’évolution des
processus cancéreux (figure 1) [5, 11].
En 2002, Cobbs et al. ont proposé des techniques sen-
sibles d’immuno-histochimie, d’hybridation in situ et de
PCR nichée capables de détecter de faibles quantités de
protéines ou de génome du HCMV dans les tissus tumo-
raux [12]. Ces techniques ont permis de détecter le HCMV
spécifiquement dans les tissus tumoraux mais non dans les
tissus sains dans plusieurs cancers comme les gliomes, les
cancers du côlon, de la prostate, de la peau, du sein, des
glandes salivaires ou encore dans les médulloblastomes
(tableau 1) [12-23]. Notre groupe a également rapporté la
présence de HCMV dans les carcinomes hépatocellulaires
(CHC) ainsi qu’une séroprévalence plus élevée du HCMV
chez des patients cirrhotiques atteints de CHC (74 %) que
chez des patients cirrhotiques indemnes de CHC (54 %)
[24]. Dans les cancers étudiés, le niveau d’expression pro-
téique ou génomique du HCMV était faible, nécessitant
l’emploi de techniques suffisamment sensibles pour détec-
ter le virus [25]. Le mode de réplication du HCMV sur le site
tumoral, prolongé et à bas bruit, fut qualifié par Söderberg-
316 Virologie, Vol 16, n◦5, septembre-octobre 2012
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

Journal Identification = VIR Article Identification = 0462 Date: October 19, 2012 Time: 1:45 pm
revue
M
S
G1
G0
G2
Monocytes porteurs
du HCMV latent
LATENCE
Macrophage
Différenciation Réplication du HCMV
Libération des
virions
??
Cellule
immune Cellule
stromale
Cellule
tumorale
FOYER INFLAMMATOIRE
Inflammation
Survie et prolifération cellulaires
Inhibition de l'apoptose
Stimulation de l’angiogenèse
Échappement immunitaire
Instabilite génétique
Invasivité
Figure 1. Modèle de l’effet oncomodulateur du cytomégalovirus humain (HCMV) dans les processus tumoraux. Les monocytes
circulants constituent un des sites de latence du HCMV. En contexte inflammatoire, la différenciation des monocytes en macrophages
s’accompagne d’une réactivation du HCMV. Le HCMV véhiculé au cœur du processus inflammatoire par les monocytes pourrait infecter
les cellules permissives locales : cellules tumorales, immunitaires ou stromales. L’influence des protéines du HCMV sur l’inflammation,
la survie, l’apoptose, l’angiogenèse, l’instabilité génétique, l’invasivité ou encore l’échappement immunitaire pourrait ainsi favoriser le
développement tumoral.
Naucler de « micro-infection » [5, 11]. Récemment, certains
auteurs ont montré que le niveau d’expression du HCMV
dans les cancers était en fait variable d’un individu à l’autre
et pouvait parfois être corrélé à la sévérité de la maladie.
Ainsi, la quantification du HCMV dans les gliomes par
une technique de PCR en temps réel a révélé des charges
virales faibles mais différentes selon les patients (102-
106copies/500 ng d’ADN total) [13]. De plus, le niveau
d’expression des protéines du HCMV dans les gliomes
était étroitement lié au grade de la tumeur [17]. En étu-
diant l’implication du HCMV dans les cancers des glandes
salivaires, Melnick et al. ont également noté une corrélation
entre le niveau d’expression des protéines du HCMV et le
grade tumoral [23]. Dans cette étude, les auteurs se sont en
outre attachés à montrer que :
– le HCMV était présent dans une proportion élevée de
tumeurs (97 %) ;
– le virus était absent dans les tissus non tumoraux ;
– l’expression des protéines du HCMV était accompagnée
d’une surexpression de voies de signalisation oncogènes
dans les tissus tumoraux ;
– le cytomégalovirus (CMV) induisait une transfor-
mation des cellules de glandes salivaires murines in
vitro.
Grâce à ces propriétés, les auteurs ont conclu que le
CMV devait être considéré comme un « oncovirus »
[23].
Au sein des tissus tumoraux, le HCMV a été détecté à
la fois dans les cellules cancéreuses et dans les cellules
du microenvironnement tumoral comme les cellules
immunitaires infiltrantes et les cellules stromales (dans les
cancers du sein et du côlon), et les cellules microgliales
(dans les gliomes) [7, 14, 20]. L’effet du HCMV sur la
tumeur ne doit donc pas être envisagé uniquement sous
Virologie, Vol 16, n◦5, septembre-octobre 2012 317
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

Journal Identification = VIR Article Identification = 0462 Date: October 19, 2012 Time: 1:45 pm
revue
Tableau 1 Détection du cytomégalovirus humain (HCMV) dans les cancers.
Type de tumeur Méthodologie Nombre de tumeurs
+ / nombre de
tumeurs étudiées
Protéines / gènes
cibles
Remarques Référence
Gliomes
Glioblastomes,
oligoastrocytomes
grade III, astrocytomes
grade II
IHC, HIS •27/27 (100 %) •IE1-72 Absence de HCMV dans les
méningiomes, Alzheimer, AVC,
encéphalites et chez les patients
sans pathologie du SNC
[12]
•10/10 •pp65
•10/10 •p52/76 kD IE/EA
PCR nichée •7/9 •UL 55
Glioblastome multiforme IHC •8/49 (16 %) •IE1-72 [19]
•25/49 (51 %) •pp65
Glioblastome multiforme IHC, HIS •42/45 (93 %) •IE1-72 Absence de HCMV en zone
saine, dans les méningiomes
et les épendymomes
[15]
•30/33 (91 %) •pp65
PCR nichée •21/34 (62 %) •UL 55
Glioblastomes, gliomes
anaplasiques, gliomes de
bas grade
IHC, HIS •44/50 (88 %) •IE1-72 Absence de HCMV en
zone non tumorale
Corrélation entre niveau
d’expression de IE1 et
grade tumoral
[17]
Gliomes PCR nichée •16/17 (94 %) •UL 123 Quantification possible par
qPCR dans 37,5 % des
cas : charges faibles et
variables
Séquenc¸age viral
retrouvant une grande
conservation des régions
US et UL
[13]
Immunoblotting •9/13 (75 %) •IE, pp65
Cancers colorectaux
Polypes colorectaux IHC, HIS •14/17 (82 %) •IE1-72 Absence de HCMV en zone saine
Colocalisation de HCMV avec
Cox-2
[14]
•7/9 (78 %) •pp65
PCR nichée •1/1 •UL 55
Adénocarcinomes IHC, HIS •12/15 (80 %) •IE1-72 Absence de HCMV en zone saine
Colocalisation de HCMV avec
Cox-2
[14]
•11/12 (92 %) •pp65
PCR nichée •6/6 •UL 55
Cancers de la prostate
Hyperplasie des cellules
basales, néoplasie
intraépithéliale prostatique,
carcinome prostatique
IHC, HIS •20/20 (100 %) •IE1-72 Absence de HCMV en
zone saine
[16]
•10/13 (77 %) •pp65
PCR nichée •9/10 •UL 73
Cancers de la peau
Carcinomes épidermoïdes PCR 8/24 (33 %) •UL 123
Absence de détection des autres
virus HSV-1/2 et EBV sur le site
tumoral
[18]
Carcinomes
basocellulaires
PCR 27/72 (37,5 %) •UL 123 [18]
Maladie de Bowen PCR 1/5 (20 %) •UL 123 [18]
Lésions prénéoplasiques PCR 6/8 (75 %) •UL 123 [18]
318 Virologie, Vol 16, n◦5, septembre-octobre 2012
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

Journal Identification = VIR Article Identification = 0462 Date: October 19, 2012 Time: 1:45 pm
revue
Tableau 1. (Suite)
Type de tumeur Méthodologie Nombre de tumeurs
+ / nombre de tumeurs
étudiées
Protéines / gènes
cibles
Remarques Référence
Cancers des glandes salivaires
Carcinome
muco-épidermoïde des
glandes salivaires
IHC •38/39 •IE1-72, pp65 Absence de HCMV en zone
saine
Colocalisation de HCMV
avec Cox-2, amphiréguline,
EGFR, ERK
Corrélation entre expression
du HCMV et grade tumoral
[23]
Médulloblastomes
Médulloblastomes IHC, IFI, HIS,
cytométrie de
flux
•34/37 (92 %) •Protéines très
précoces (IE)
Corrélation entre
expressions de HCMV et
Cox-2
Croissance tumorale
diminuée en présence de
ganciclovir ou d’inhibiteur
de Cox-2
[22]
•27/37 (73 %) •Protéines
tardives
PCR •6/6 •IE, UL 32
(pp150)
Cancers du sein
Sein IHC, HIS •31/32 (97 %) •IE1/2 Détection dans l’épithélium
glandulaire non tumoral
dans 63 % des cas avec
des anticorps dirigés
contre les antigènes IE et
21 % des cas avec des
anticorps contre les
antigènes E/L
[20]
•21/25 (84 %) •E, L
PCR nichée •6/8 •UL 55
Cancers du poumon
Poumon PCR nichée •18/78 (23 %) Antigène
précoce
Absence de HCMV dans
les tissus pulmonaires
sains
[21]
IHC : immuno-histochimie ; HIS : hybridation in situ ; IFI : immunofluorescence indirecte ; PCR : réaction d’amplification génique ; qPCR : réaction de PCR
quantitative ; IE : protéines très précoces ou immediate early ; E : protéines précoces ou early ; L : protéines tardives ou late ; EBV : virus d’Epstein-Barr ;
Cox-2 : cyclo-oxygénase 2.
l’angle de la « simple » interaction entre le virus et la
cellule cancéreuse, mais plutôt dans une perspective plus
large d’interactions multiples entre le virus et l’ensemble
des cellules présentes dans la tumeur, qu’elles soient
tumorales, stromales ou immunitaires (figure 1). Nous
développerons donc dans les paragraphes suivants à la
fois les effets « oncomodulateurs » du HCMV sur le
microenvironnement tumoral et sur le système immuni-
taire, et son effet protumoral sur la cellule cancéreuse ou
précancéreuse.
Action du cytomégalovirus humain
sur le microenvironnement tumoral
L’infection par le cytomégalovirus humain
induit une inflammation
L’interaction entre les cellules cancéreuses et le micro-
environnement tumoral est essentielle au développement
et à la croissance de la tumeur. Le microenvironnement
tumoral est le siège d’une inflammation chronique qui
Virologie, Vol 16, n◦5, septembre-octobre 2012 319
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.
 6
7
8
9
10
11
12
13
14
15
6
7
8
9
10
11
12
13
14
15
1
/
15
100%