application à la pathologie

BASES MOLECULAIRES ET CELLULAIRES DES PATHOLOGIES- PARTIE I - Régulation de l'expression génique :
application à la pathologie
22/10/2013
WINNICKI Camille L2
BMCP
Pr A. Margotat
16 pages
Régulation de l'expression génique : application à la pathologie
PARTIE I
A. Introduction
Le génome humain est composé d'environ 25000 gènes (Gène dans le sens classique c'est-à-dire une
séquence précise d'ADN exprimant une protéine)
L'expression de chaque gène doit être contrôlée dans le temps et l'espace c'est-à-dire à l'endroit voulu et au
moment voulu.
Lorsqu'on parle d'un gène de façon classique (codant pour une protéine), on a
→ une séquence d'ADN
→ Le commencement par un promoteur (région dans laquelle va commencer la transcription) + un certain
nombre d'exons (partie codante) et d'introns (éliminés au cours de la maturation/épissage).
→ dans les premiers et le dernier exons, il existe des régions non traduites (en 3' et 5') ainsi qu'un site de
polymérisation.
En moyenne :
la région 5'UTR en région promotrice : 200 à 300 pb (soit 0,2 à 0,3 kb)
La région 3'UTR en région terminale : 700 à 800 pb (soit 0,7 à 0,8 kb)
La taille des introns subit d'énormes variations de quelques dizaines à quelques centaines de pb.
Exemples :
β-globine : 2kb et 3 exons
BRCA 1 : 80 kb et 24 exons
NYH7 : 20kb et 40 exons
1/16
Plan :
A. Introduction
B. Zones de régulation de l'expression génique
C. Remodelage de la chromatine
D. Modifications post traductionnelles des histones
I. Acétylation
II. Méthylation
III. Phosphorylation
IV. Ubiquitinylation
V. Désacétylation
E. Méthylation de l'ADN
F. Héritage de générations en générations
G. Eléments « cis » et facteurs « trans »

BASES MOLECULAIRES ET CELLULAIRES DES PATHOLOGIES- PARTIE I - Régulation de l'expression génique :
application à la pathologie
La taille d'un exon est relativement homogène et est en moyenne 122 pb (variations entre 100 et 200 pb)
Le nombre moyen d'exons est de 9 par gène mais subit de considérables variations.
Exemples :
•Taille moyenne d'un ARNm : 2,6 kb (considérables variations entre les ARNm)
alors que :
•La titine : 115 kb
•La taille moyenne d'un gène humain ( plus difficile à connaitre car exons, introns, promoteur
proximal...) : 27 kb
Si on considère la médiane, elle est de 14 kb ( certains gènes sont très longs, ex : dystrophine = 2400kb)
Si la moyenne est différente de la médiane, on assiste à une distribution pas normale dans le sens mathématique
On estime qu'environ 4% de la longueur d'un gène humain est codante pour une protéine. 1,5% du génome est
donc codant et 25% représente des gènes codant pour des protéines.
Pour illustrer la très grande diversité de taille des gènes, on peut distinguer 3 « catégories » :
1. Gènes de moins de 10kb
•gènes d'histones ( - d'1kb) : 100% de la séquence est codante ( pas d'introns mais uniquement des exons)
•gène de la β-globine : 38% exons
2.Gènes entre 10 et 100kb :
•le pourcentage diminue de 12 à 3% pour la phénylalanine par exemple. Ainsi lorsqu'un gène s'agrandit,
il s'agrandit par le nombre et la taille de ses introns alors que la taille des exons reste relativement
constante. Par conséquent, le pourcentage de codons diminue.
3. Gène de taille > 100kb
Gène de la dystrophine (mutation → myopathie de Duchenne)
On observe une extrême variabilité des échelles de taille.
Rappel : Diapo illustrant la transcription puis la traduction d'un gène en protéine.
2/16

BASES MOLECULAIRES ET CELLULAIRES DES PATHOLOGIES- PARTIE I - Régulation de l'expression génique :
application à la pathologie
B. Zones de régulations
Toutes les étapes de transcription peuvent être un site de régulation.
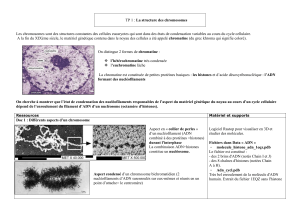
La photo n'étant pas nette même sur le diaporama, je suis désolée de la qualité de la photo... Mieux vaut aller
voir directement sur l'ENT...
1ère zone de régulation : la chromatine elle-même (ensemble d'ADN et de protéines dont les histones)
( /!\ différent de l'ADN ou des histones seuls )
La chromatine correspond au génome fonctionnel pour laquelle il existe de nombreuses régulations possibles :
1. l'assemblage entre ADN et nucléosome ( c'est-à-dire les protéines histones ) peut être modifié à
tout moment. La modification post-traductionnelle des histones modifie la structure de cette
chromatine.
2. La méthylation des cytosines de l'ADN qui va lui aussi réguler l'accès d'autres protéines
régulatrices à l'ADN pour faire s'exprimer/réprimer l'expression des gènes.
2ème zone au niveau transcriptionnel : sites CYS (séquences d'ADN particulières au voisinage des gènes)
1. Cette séquence accueille des protéines régulatrices appelées effecteurs TRANS qui sont des protéines
qui vont reconnaître une séquence d'ADN CYS pour réguler l'expression des gènes.
2. On peut réguler également l'expression des gènes en choisissant des promoteurs différents pour un
même gène.
3. La maturation de l'ARN est également un site de régulation via l'épissage alternatif : choix de
différents exons.
3ème zone : niveau post-transcriptionnel
•Si la polyadénylation ne se fait pas correctement, la durée de vie de l'ARNm sera altérée.
•De même, au cours de son transport, l'ARNm peut être dégradé avant d’être transporté.
•L' interférence ARN : régulation de la traduction par des ARN non codants.
3/16

BASES MOLECULAIRES ET CELLULAIRES DES PATHOLOGIES- PARTIE I - Régulation de l'expression génique :
application à la pathologie
On ne parlera pas ici de la régulation purement traductionnelle (au niveau des ribosomes).
4ème niveau : niveau post-traductionnel
La maturation post-traductionnelle des protéines représente autant de site de régulations.
ex : protéine non glycosylée → incapable de remplir sa fonction.
Lorsqu'on parle d'un gène et de sa régulation, tous les éléments clés de la régulation de la transcription
(éléments régulateurs proximaux, distants, introns, exons, etc.) sont inclus dans une structure tridimensionnelle
où l'ADN est entouré autour des histones.
Il faut que l'ADN polymérase puisse naviguer dans cette structure tridimensionnelle afin d'assurer la
transcription.
C) Remodelage de la chromatine:
Les modifications de la chromatine régulent l'expression du génome :
Physiquement, entre les divisions cellulaires, l'ADN se retrouve sous la forme de chromatine présente dans le
noyau. Selon la phase de la division cellulaire on va pouvoir ± observer cette chromatine. Celle-ci présente des
plages sombres ou claires.
Chromatine = ADN enroulé autour de protéines Histones + présence de protéines non Histones.
Si on regarde la chromatine de façon plus précise on remarque deux types de chromatine :
•Région assez claires : l'euchromatine → partie du génome impliquée dans l'expression des gènes. C'est
la partie active du génome. Elle est dispersée dans le noyau.
•Région sombre en MO : l'hétérochromatine à la périphérie du noyau → région du génome qui est
impliquée dans la structure, la compaction dans les divisions. Elle ne contient pratiquement pas de gènes
exprimés. C'est donc une région inactive du point de vue de la transcription.
On considère deux formes différentes d'hétérochromatine :
→ Constitutive : +++ compactée et située à l'extrémité vers les télomères.
Sa séquence contient de nombreuses répétitions nécessaires à la réplication des télomères (aucun intérêt pour
les gènes mais dans la conservation de la même longueur des chromosomes au fil des divisions). Elle est riche
en nucléotides A et T.
→ Facultative : Selon les moments du cycle, elle peut se décompacter et produire quelques ARN
(dont rôle parfois inconnu). Ces régions contiennent de nombreuses séquences répétitives de type LIME
susceptibles de transposer d'un endroit à l'autre du génome. Il existe des périodes de la vie cellulaire où cette
chromatine-là se décondense.
Si on considère l'euchromatine, elle peut se trouver sous deux formes:
•Filaments : de 11-12 nanomètres entourés autour de nucléosomes. C'est une forme détendue.
•Forme inactive : Sous forme de solénoïde ++ compactée de 30 nm.
Au moment des divisions cellulaires il y une compaction de 6000 fois la longueur de chaque chromosome :
cela implique 2 nouveaux états de repliement qui recrutent d'autres protéines que les histones.
4/16

BASES MOLECULAIRES ET CELLULAIRES DES PATHOLOGIES- PARTIE I - Régulation de l'expression génique :
application à la pathologie
D. Modifications post-traductionnelles des histones:
NUCLEOSOMES :
La structure de base de la chromatine est d'environ 147pb autour d'un nucléosome, et la présence d'un
nucléosome tous les 200/300 pb.
Ce sont des octamères protéiques compacts de type H2A, H2B, H3, H4.
Ce sont des petites protéines très conservées au fil des ans, qui sont codées par des gènes sans introns.
Elles sont codées par des gènes qui ne possèdent pas d'introns car cela permet une production rapide.
Lorsque ces histones sont associées avec l'ADN, l'extrémité N-terminale des histones est saillante à l'extérieur
du nucléosome. Ces mêmes extrémités N-terminales peuvent être modifiées.
Au pH cellulaire, elles sont chargées positivement car elles sont riches en acides aminés basiques (arginine,
lysine) → elles vont interagir avec le squelette de l'ADN qui lui est chargé négativement et assurer la stabilité
de l'ADN.
Elles peuvent être modifiées de façon transitoire par modification de leur charge. Cela permet la modification
de la structure de la chromatine (au moment de la réplication et de la transcription) en modifiant la charge donc
en modifiant l'interaction avec les phosphates (chargés négativement) de l'ADN.
Exemple : l'Histone H1 (qui ne fait pas partie du nucléosome) peut être phosphorylée.
Modifications post-traductionnelles des histones :
3 exemples :
•Phosphorylation : Sur les sérines grâce à des kinases/phosphatases.
•Méthylation : Sur les lysines ou arginines. Grâce à des enzymes, les histones méthylases ( HMT), et
Déméthylation grâce à une seule déméthylase connue : la LSD1.
•Acétylation : Sur les lysines, grâce à des histones acétyl-transférases (HAT), et Désacétylation grâce à
des histones désacétylases (HDAC).
•Ubiquitinylation : sur des lysines grâce à des enzymes E1,E2,E3/isopeptidase.
Chacune de ces modifications va diminuer la charge et donc diminuer la force de l'interaction des histones avec
l'ADN.
5/16
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%