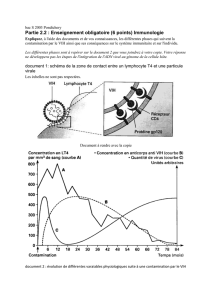
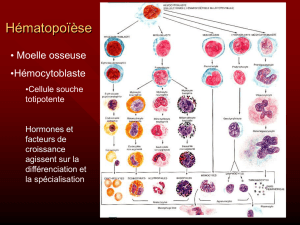
71852_MUSILLI_2016_archivage.p... - Tel Archives ouvertes

Etude des m´ecanismes d’action du Strontium 90 sur le
syst`eme immunitaire `a la suite d’une contamination
chronique
Stefania Musilli
To cite this version:
Stefania Musilli. Etude des m´ecanismes d’action du Strontium 90 sur le syst`eme immunitaire
`a la suite d’une contamination chronique. Toxicologie et chaˆıne alimentaire. Universit´e Paris-
Saclay, 2016. Fran¸cais. <NNT : 2016SACLS077>.<tel-01499151>
HAL Id: tel-01499151
https://tel.archives-ouvertes.fr/tel-01499151
Submitted on 31 Mar 2017
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-
entific research documents, whether they are pub-
lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destin´ee au d´epˆot et `a la diffusion de documents
scientifiques de niveau recherche, publi´es ou non,
´emanant des ´etablissements d’enseignement et de
recherche fran¸cais ou ´etrangers, des laboratoires
publics ou priv´es.

NNT : 2016SACLS077
THESE DE DOCTORAT
DE
L’UNIVERSITE PARIS-SACLAY
PREPAREE A
L’INSTITUT DE RADIOPROTECTION ET DE SURETE NUCLEAIRE
ECOLE DOCTORALE N° 569 ITFA
Innovation Thérapeutique : du fondamental à l’appliqué
Toxicologie
Par
Stefania Musilli
Etude des mécanismes d’action du Strontium 90 sur le système immunitaire à la suite
d’une contamination chronique
Thèse présentée et soutenue à l’IRSN, Fontenay-aux-roses le 30 Mars 2016 :
Composition du Jury :
Pr. Armelle Baeza-Squiban
Université Paris Diderot
Rapporteur
Dr. Francis Herodin
Institut de recherche biomédicale des armées (IRBA)
Rapporteur
Pr Chantal Houée-Levin
Université Paris Sud
Présidente du jury
Pr. Xavier Coumoul
Université Paris Descartes
Examinateur
Pr. Saadia Kerdine-Römer
Université Paris Sud
Co-directrice de thèse
Dr. Jean-Marc Bertho
Institut de radioprotection et de sureté nucléaire (IRSN)
Directeur de thèse

2

3
Si haute est la montagne, n’oublie jamais qu’il y a toujours
un chemin qui mène au sommet - Proverbe afghan

4
Remerciements
Je tiens tout d’abord à remercier les membres du jury pour l’évaluation de ce travail. Un grand
merci aux rapporteurs, le Dr. Francis Herodin et le Pr. Armelle Baeza pour le temps que vous
avez accordé à la relecture de ce manuscrit. Je remercie également le Pr. Chantal Houée-Levin et
le Pr. Xavier Coumoul pour leur présence en tant qu’examinateurs dans ce jury.
Je remercie également les personnes de l’Institut de Radioprotection et de Sûreté Nucléaire sans
qui, ce travail n’aurait été possible. Merci au Dr. Jocelyne Aigueperse pour m’avoir permis de
réaliser ma thèse au sein du Pôle de Radioprotection de l’Homme ainsi qu’au Dr. Marc
Benderitter et au Dr. Laurence Roy du Service de radiobiologie et d’Epidémiologie. Enfin, un
grand merci au Dr. Karine Tack pour son accueil au sein du Laboratoire de Radiotoxicologie
expérimentale.
Je remercie chaleureusement le Pr. Saadia Kerdine-Römer pour avoir co-encadré cette thèse.
Merci pour les échanges scientifiques toujours très enrichissants que nous avons eu au cours de
ces 3 années et pour tes précieux conseils. Ce n’était pas toujours facile de trouver des
disponibilités pour se voir mais on y sera arrivées au moins pour la date la plus importante. Un
grand merci pour tout.
Un immense merci au Dr. Jean-Marc Bertho pour m’avoir encadrée au cours de ces 3 longues
années et demi de thèse. Merci pour ta patience, ton écoute, tes conseils, pour toutes les
discussions scientifiques (et moins scientifiques), pour m’avoir permis de devenir une pro des
ELISA et surtout pour la confiance que tu m’as accordée dès le M1. Pour tout ça, encore merci !
Mes remerciements vont également vers l’équipe technique de choc du LRTOX.
Merci à Line Manens pour tous les bons moments que nous avons passé ensemble, pour les
discussions toujours très passionnantes et surtout pour le désormais célèbre Vendredi paillettes
qui, j’en suis sûre, va bientôt devenir une mode au sein de tout le SRBE.
Un grand merci aussi à Céline Gloaguen pour ta bonne humeur, ton calme à toute épreuve, pour
ton super biceps de compet. C’était vraiment un plaisir de partager cette dernière année avec toi.
Je tiens aussi à remercier Dimitri Kerdlsi…Kerselzd…Kerdlizds…Kereselidze pour les supers
mercredi midi sportifs, pour ne m’avoir jamais mis de bâtons dans les roues et pour avoir eu la
patience de supporter les nombreuses blagues sur les chauves !
Mes remerciements les plus chaleureux vont à Christelle Elie. Merci pour tous les bons moments
partagés ensemble, pour avoir été là quand j’en avais besoin, pour m’avoir supportée dans les
bonnes et les moins bonnes périodes, surtout au cours de ces derniers mois ! Merci pour avoir
écouté toutes mes divagations, tu as été mon meilleur public depuis nos débuts ensemble au
LRTOX il y a 3 ans (finalement cette thèse c’est un peu comme si tu l’avais faite aussi ). Pour
tout ça un très grand merci !!!
Et évidemment je n’oublie pas de remercier Yohann Phalente, c’était un plaisir de connaître un
grand monsieur comme toi.
Un grand merci aux chercheurs du LRTOX. Merci au Dr. Philippe Lestaevel pour tes
encouragements surtout dans cette dernière ligne droite, pour les bons moments passés à Porto
(un peu de poulpe ?) et pour toutes les discussions et conseils au cours de ces années. Un grand
merci aussi au Dr. Maâmar Souidi pour ses nombreux encouragements et ses conseils. Mes
remerciements vont également aux Dr. Téni Ebrahimian, Yann Guéguen, Chrystelle Ibanez et
Audrey Legendre pour leur support et les discussions horoscope du midi.
Un énorme merci au Dr. Christelle Durand pour toutes nos conversations, pour tes conseils, ton
écoute et pour ta voiture . Tu n’étais qu’une jeune thésarde quand on s’est connues et j’ai
vraiment été heureuse de te retrouver en tant que chercheuse et surtout qu’on apprenne à se
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
1
/
227
100%