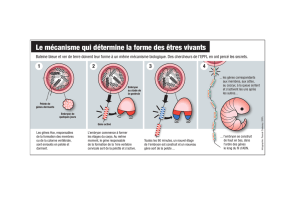
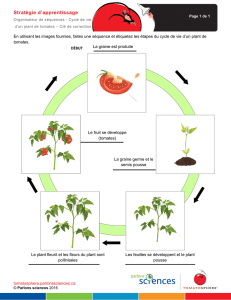
Contribution de l`albumen au développement de la graine

UNIVERSITE DE BOURGOGNE
Ecole Doctorale Environnements Santé n°554
Institut National de la Recherche Agronomique
UMR 1347 Agroécologie - pôle GEAPSI
THÈSE
Pour l'obtention du grade de
Discipline : Sciences Vie
Spécialité: Biologie moléculaire végétale
C
chez Medicago truncatula
par Mélanie NOGUERO
soutenue le 23 Mai 2014
Gwyneth INGRAM, Chargée de recherches, CNRS, ENS Lyon Rapporteur
Loic RAJJOU, Maître de conférences, AgroParisTech Rapporteur
Nahalie LEBORGNE-CASTEL, Professeur Universités, Dijon Examinateur
Bertrand DUBREUCQ, Directeur de recherches, INRA, AgroParisTech Examinateur
Karine GALLARDO, Chargée de recherches, INRA, Dijon Co-encadrant de thèse
Richard THOMPSON, Directeur de recherches, INRA, Dijon Directeur de thèse


Résumé
Dans un contexte actuel qui tend vers une r
relance de la production de protéines végétales pour réduire la dépendance alimentaire de la France, la
culture des légumineuses constitue une alternative. Les légumineuses à graines offrent une source riche en
Au sein de l'UMR1347 Agroécologie, le pôle
Innovants" (GEAPSI) étudie via une approche multidisciplinaire (génétique, écophysiologie, physiologie
environnementales. Ces travaux de thèse ont été réalisés au sein de l'équipe "Étude des Mécanismes
Moléculaires" qui s'intéresse plus particulièrement à des critères de qualité de la graine (teneur en
protéine, taille de graine) et au déterminisme génétique de ces caractères chez la plante modèle Medicago
truncatula en vue d'un transfert de connaissances vers l'espèce cible Pisum sativum.
Aux stades précoces du
e nutriments est réalisée majoritairement par les tissus qui entourent
dans le développement de la graine chez M. truncatula. Nous avons révélé plusieurs gènes DOF (DNA-
binding with One zinc Finger) comme étant exprimé dans ce tissu. Ils appartiennent à une large famille de
facteurs de transcription impliqués dans de nombreux processus biologiques mais dont les rôles restent à
préciser. Un de ces gènes, nommé DASH pour Dof Affecting Seed embryogenesis and Hormone
metabolism, est exprimé . Des mutants TILLING
et Tnt-1 isolés pour ce facteur de transcription sont affectés dans le développement de la graine
(avortement à 10 jap). La cytologie du développement aux stades précoces (6 à 10 jap) a révélé que
,
ryogenèse chez les légumineuses. Une étude
comparative du transcriptome des gousses de dash vs sauvage émettre des hypothèses quant à
la fonction du gène DASH. Une
mise en évidence et plusieurs gènes cibles potentiels de ce facteur de transcription ont été sélectionnés.
Une comparaison du transcriptome des trois tissus de la graine à 12 jap a été réalisée chez la lignée
sauvage de référence (Jemalong, A17). Elle a permis de préciser la localisation tissulaire de ces gènes cibles
putatifs, de mettre en évidence des voies métaboliques plus spécifiques et de proposer des
hypothèses quant à la fonction de ce tissu dans le développement de la graine.
Mots-clés: Développement de la graine, Medicago truncatula, caractérisation de mutants, étude
comparative du transcriptome, analyse cytologique.

Abstract
In the current context, which necessitates a reduction in inputs in crop systems and boosting of
production of plant proteins to reduce dependency on feed imports,, growing legumes represents
an alternative. Grain legumes are major sources of proteins for animal and human nutrition. In the
UMR1347 Agroécologie, the objectives of the study group "déterminismes Génétiques et
promote legume cultivation and adaptation to environmental stresses, via multidisciplinary approaches
(genetics, ecophysiology, molecular physiology). This thesis project was carried out in the "Étude des
Mécanismes Moléculaires" team, particularly interested in seed quality traits such as protein content or
seed size and identification of genes implicated in variations of these trait s. Experiments were performed
using Medicago truncatula as a model species for legumes with a view to transferring the information to
the target crop species Pisum sativum.
accumulate reserves during seed filling. At early developmental stages, nutrient assimilation occurs
predominantly in embryo-surrounding tissues: the endosperm and seed coat. This thesis project aims to
evaluate the endosperm contribution to seed development in M. truncatula. We have shown several DOF
(DNA-binding One Zinc Finger) genes to be expressed in this tissue. They belong to a large family of
transcription factors implicated in numerous biological processes, but whose role remain to be elucidated.
One of these genes, termed DASH for Dof Affecting Seed embryogenesis and Hormone metabolism, is
expressed preferentially in the endosperm during embryogenesis. TILLING and TnT1 mutants isolated for
this transcription factor are affected in seed development (abortion at 10 DAP). The cytology of
development at early stages (6 to 10 DAP) revealed that the expression of this gene in the endosperm is
required for the normal development of the embryo, demonstrating the role of the endosperm in the
control of embryogenesis in legumes. A comparative transcriptomics study of dash vs wild-type pods
allowed us to suggest hypothesis about the function of the DASH gene. Evidence for a deregulation of
hormone metabolism, in particular for auxin, was obtained, and several potential target genes of this
transcription factor were selected. A comparison of the transcriptome of the three tissues of the seed at 12
DAP was carried out for the reference wild-type line (Jemalong A17). This allowed the tissue localization of
the target genes, to reveal metabolic pathways preferentially expressed in the endosperm, and to propose
hypotheses about the role of this tissue during seed development.
Keywords: seed development, Medicago truncatula, mutant characterisation, transcriptomic analyses,
cytological studies.

Liste des abbréviations
ABA Acide ABscissique
AIA/IAA Acide Indole Acétique
alb albumen
ARF Auxin Response Factor
BAC Bacterial Artificial Chromosome
bHLH basic Helix Loop Helix
BLAST Basic Local Alignment Search Tool
CGDD Commissariat Général au Développement Durable
dap days after polination
DASH DOF Acting in Seed embryogenesis and Hormone regulation
DOF DNA binding with One Finger
emb embryon
EMS Ethyl methanesulfonate
ERF Ethylene Response Factor
EST Expressed Sequence Tag
FT Facteur de Transcription
GES Gaz à Effet de Serre
jap jours après polinisation
LEA Late Embryogenesis Abundant
MtGEA Medicago truncatula Gene Expression Atlas
NLS Nuclear Localization Signal
PAC Politique Agricole Commune
q-PCR PCR quantitative
QTL Quantitative Trait Loci
teg tégument
TAIR The Arabidopsis Information Resource
TIBA 2,3,5-triiodobenzoic acid
TF Transcription Factor
TILLING Targeting Induced Local Lesions IN Genome
UNIP Union Nationale Interprofessionnelle des plantes riches en Protéines
WT Wild-type/ génotype sauvage
2-4D acide 2,4-dichlorophénoxyacétique
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
1
/
185
100%