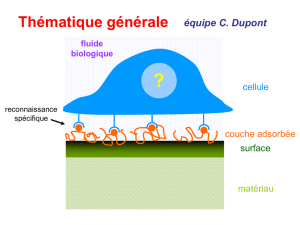
THÈSE DOCTEUR EN CHIMIE

REPUBLIQUE ALGERIENNE DEMOCRATIQUE POPULAIRE
MINISTERE DE L’ENSEIGNEMENT SUPERIEUR
ET DE LA RECHERCHE SCIENTIFIQUE
UNIVERSITE ABOU–BAKR BELKAID –TLEMCEN
FACULTÉ DES SCIENCES
DEPARTEMENT DE CHIMIE
Organiques
(LAEPO)
THÈSE
Pour obtenir le grade de
DOCTEUR EN CHIMIE
Spécialité : Chimie et Physico-chimie Organique Macromoléculaire
Par
Mme Soraya BELKAID ép. BOUHADJER
Sur le Thème
Copolymères P4VP-bromure d’hexadécyle, Caractérisation physico-
chimique, Propriétés inhibitrices et Immobilisation sur la bentonite –
Application à la rétention des polluants organiques
Soutenue publiquement en juillet 2013, devant le jury composé de:
Président: Pr. Said GHALEM (Univ. Tlemcen)
Directeur de Thèse Pr. Ali MANSRI (Univ. Tlelemcen)
Examinateurs: Pr. Soufi KACIMI (Centre Univ. Ain-Temouchent)
M.C.A. Lahcen BELARBI (Centre Univ. Ain-Temouchent)
M.C.A. Omar BENALI (Univ. Saida)
Pr. Boufeldja TABTI (Univ. Tlemcen)
Invité: M.C.A. Nesr-Eddine BELKHOUCHE (Univ. Tlemcen)

Remerciements
C’est avec un réel plaisir et un grand enthousiasme que je me livre à la
rédaction de cette page. Bien plus que le point final de la rédaction scientifique, cette
page représente une méditation sur une période relatant six années de vie très riches
en évènements.
Ce travail a été effectué au Laboratoire d’Application des Electrolytes et des
Polyélectrolyte Organiques (LAEPO) de l’Université Abou Bakr-Belkaid de Tlemcen,
dirigé par Monsieur le Professeur Ali Mansri.
Je tiens tout d’abord à exprimer mon profond respect et mes remerciements les
plus sincères au professeur Ali Mansri. Je lui suis profondément reconnaissante de
ses conseils, ses encouragements. Son expérience et son tempérament chaleureux ont
été pour moi une source permanente d’enrichissement.
Je suis très reconnaissante à Monsieur Bruno Grassel, Directeur de l’équipe
chimie à l’Institut Pluridisciplinaire de Recherche sur l’Environnement et les
Matériaux (IPREM) à Pau (France), qui nous a permis d’effectuer les analyses de
RMN et ATG au sein de son laboratoire.
Mes remerciements s’adressent également à Monsieur Belkheir Hammouti de
nous avoir permis d’effectuer les mesures électrochimiques de la corrosion à
l’Université Mohammed Premier à Oujda (Maroc).
Que Monsieur Said Ghalem professeur à l’Université de Tlemcen qui a bien
voulu me faire l’honneur de juger ce travail et de présider mon jury de thèse de
doctorat. Je lui adresse mes remerciements les plus respectueux.

Je voudrais remercier également Messieurs Soufi Kacimi Professeur au Centre
Universitaire de Ain-Temouchent, Lahcen Belarbi Maître de Conférences A au Centre
Universitaire de Ain-Temouchent, Omar Benali Maître de Conférences A à
l’Université de Saida, Boufeldja Tabti Professeur à l’Université de Tlemcen pour avoir
mobilisé de leurs temps pour l’intérêt qu’ils ont porté à mon travail et pour avoir
accepté de l’examiner.
Je remercie Mr Nasr Eddine Belkhouche Maître de Conférences A à
l’Université de Tlemcen d’avoir accepter de participer à ce jury.
Je remercie également Madame Esma Choukchou-Braham et Monsieurs Ismet
Benabadji, Lahcen Tannouga, pour leurs conseils et encouragements.
Je tiens à remercier également Mr Mourad Boufateh responsable technique du
laboratoire de chimie physique du département de chimie de l’université de Tlemcen
pour son aide et sa disponibilité durant la réalisation des mesures tensiométriques.
Un grand merci à tous les membres du Laboratoire (LAEPO) sans exception,
enseignants et thésards en particulier Bouras pour son savoir et ses conseils, Nassima,
Samia, Zolikha, Aicha, fayçal, wassila…. Nos discussions drôles et animées
participent grandement au délicieux souvenir que je conserve de cette période.
A mes parents, vous avez su guider mes pas, m’aider à devenir ce que je suis.
Ma joie de vivre, ma force et mes réussites c’est à vous que je les dois, merci d’être là
dans les moments de doute. Merci de me supporter et quoique je puisse vous dédier ça
ne sera jamais assez.
Il faut reconnaître que je n’en serai pas arrivé à ce point sans le soutien
constant de mon mari et la grande compréhension de mes enfants. Qu’ils m’excusent
pour tous les désagréments que j’ai pu leur causer.

Un grand merci pour mes frères et sœurs pour leurs encouragements et leur
soutien.
Enfin, je ne pourrais terminer sans remercier tous ceux qui, de près ou de loin,
Veuillez trouvez dans ce travail l’expression de ma profonde reconnaissance.
S. BELKAID ép. BOUHADJER

SOMMAIRE
INTRODUCTION GENERALE
CHAPITRE I: PRINCIPES ET TECHNIQUES EXPERIMENTALES
I.1.SPECTROSCOPIE RESONNACE MAGNETIQUE
I.2.SPECTROSCOPIE ULTRA-
I.4. ANALYSE THERMOG
CHAPITRE II: SYNTHESES ET CARACTERISATIONS DES
COPOLYMERES POLY(4-VINYPYRIDINE-BROMURE D’HEXADECYLE)
II.3.1. Préparations des copolymères poly(4-vinylpyridine-bromure
II.3.2. Estimation du taux de quaternisation de chaque prélèvement par
II.3.3. Estimation par conductimétrie du taux de quaternisation des
copolymères poly(4-vinylpyridine-
II.3.4.Caractérisations des copolymères poly(4-vinylpyridine-bromure
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
1
/
191
100%






![Biocompatibilité des polymères [Mode de compatibilité]](http://s1.studylibfr.com/store/data/004845602_1-0676c2f26aefaaf094e4d02094640c45-300x300.png)