L`échappement aux inhibiteurs de l`enzyme de conversion au cours

Actualités thérapeutiques
L’échappement
aux inhibiteurs de l’enzyme
de conversion au cours
de l’insuffisance cardiaque :
physiopathologie
et remèdes
Pierre Vladimir Ennezat
1
, Xavier Gonin
1
, Jean-Marc Aubert
1
,
Julie Darchis
1
, Jean-Luc Auffray
1
, Jean-Jacques Bauchart
1
,
Eric Van Belle
1
, Thierry LeJemtel
2
, Philippe Asseman
1
1
Urgences et soins intensifs, hôpital cardiologique, Lille, France
2
Tulane University, New Orleans, Louisiane
Le traitement par inhibiteur de l’enzyme de conversion de l’angiotensine I est une pierre
angulaire du traitement de l’insuffisance cardiaque. Malgré cette inhibition pharmacologique,
la production d’angiotensine II persiste, amenant au concept « d’échappement aux IEC ». Les
mécanismes en sont discutés dans cet article, ainsi que les implications thérapeutiques.
Mots clés :IEC, insuffisance cardiaque, échappement aux IEC
Il est désormais admis que l’activa-
tion du système rénine-angioten-
sine est un facteur déterminant dans le
pronostic de l’insuffisance cardiaque,
du post-infarctus du myocarde, mais
aussi des néphropathies diabétiques et
non diabétiques. Le système rénine-
angiotensine très probablement impli-
qué dans l’ensemble des maladies
cardio- et cérébrovasculaires.
Le blocage de l’axe rénine-
angiotensine-aldostérone par un inhi-
biteur de l’enzyme de conversion de
l’angiotensine I (IEC) est devenu une
des pierres angulaires de la thérapeu-
tique de l’insuffisance cardiaque, aux
côtés du blocage du système nerveux
sympathique, avec une réduction
relative de la mortalité totale d’envi-
ron 20 % en comparaison avec le pla-
cebo [1]. Cependant, malgré ces
avancées thérapeutiques majeures la
morbidité ainsi que la mortalité de
l’insuffisance cardiaque restent impor-
tantes. Parmi les nombreuses explica-
tions de cet échec apparent, en
d’autres termes de cet échappement
thérapeutique, la persistance de la
production d’angiotensine II sous IEC
peut être une réponse potentielle.
Il existe deux systèmes
rénine-angiotensine :
tissulaire versus circulant
Le système rénine-angiotensine
(SRA) (figure 1) est avant tout un sys-
tème physiologique (décrit sur un plan
phylogénique même chez l’anguille)
intrarénal de régulation fine de la
pression artérielle qui devient relative-
ment indépendante des apports sodés
m
t
Tirés à part : P.V. Ennezat
mt, vol. 12, n° 2, mars-avril 2006 71
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

[2]. De nombreux tissus peuvent produire de l’angioten-
sine II indépendamment de la rénine circulante, à l’instar
du cœur, du rein et des vaisseaux. Tous les composants
nécessaires du SRA (substrats, enzymes et récepteurs) sont
présents au niveau des tissus (figure 2). En outre, la pro-
duction plasmatique des angiotensines ne représente que
20 % de la production totale des angiotensines, 80 %
étant produits dans l’interstitium. L’angiotensine II tissu-
laire produite possède essentiellement un rôle de facteur
de croissance (remodelage cardiaque et vasculaire) ou de
neurotransmetteur dans le cerveau (soif, appétit pour le
sel, activation du système sympathique...).
L’angiotensinogène tissulaire est synthétisé à partir du
pré-pro-angiotensinogène et est un substrat pour la rénine
présente au niveau tissulaire mais aussi pour des processus
enzymatiques alternatifs comme les cathepsine G, tonine,
chymase [3] et Cage (chymostatin-sensitive angiotensin II
generating enzyme) [4]. Par conséquent, l’angiotensine II
tissulaire peut être produite indépendamment de l’ECA et
cette production d’angiotensine II dépendante des systè-
mes alternatifs n’est pas supprimée par les IEC. En revan-
che, dans l’espace intravasculaire, l’ECA plasmatique et
membranaire localisée sur la face luminale des cellules
endothéliales est exclusivement responsable de la produc-
tion d’angiotensine II [5].
L’importance de la production tissulaire d’angioten-
sine II indépendante de l’activité de l’ECA reste controver-
sée. Urata H et al. [3] démontraient que la chymase
pouvait être responsable de 80 % de la production
d’angiotensine II dans l’interstitium myocardique humain
[6]. Cependant, les conditions expérimentales même
modifient dramatiquement les résultats. In vivo, les chy-
mases sont essentiellement localisées dans les granules
secrétoires des mastocytes et sont naturellement inhibées
lorsqu’elles sont libérées, par de fortes concentrations
d’inhibiteurs de protéases localisées dans le liquide inters-
titiel. In vitro, après homogénéisation des tissus, les chy-
mases sont libérées dans le milieu expérimental et leur
activité est alors surestimée [7, 8].
Système rénine-angiotensine
et insuffisance cardiaque
En réponse à une baisse du débit cardiaque, le SRAA
s’active et interagit étroitement avec le système sympathi-
que. En effet, la stimulation des récepteurs b
1
de l’appareil
juxtaglomérulaire, conséquence de l’augmentation de
l’activité adrénergique en période de décompensation de
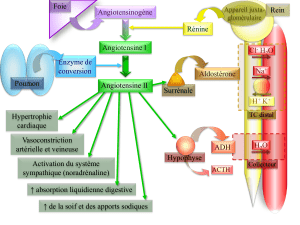
Figure 1. Le système rénine-angiotensine (SRA).
Actualités thérapeutiques
mt, vol. 12, n° 2, mars-avril 2006
72
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

l’insuffisance cardiaque, aboutit à un relargage important
de rénine dans la circulation. L’activation des barorécep-
teurs vasculaires rénaux sensibles à la baisse de pression
de perfusion rénale (conséquence de la baisse du débit
cardiaque, du régime appauvri en sel et des traitements
diurétiques) ainsi que la baisse de la quantité de sodium
filtré parvenant au niveau de la macula densa contribuent
également à la sécrétion de rénine observée dans l’insuf-
fisance cardiaque. Cette élévation de l’activité rénine plas-
matique aboutit à une augmentation de la production
d’angiotensine II avec pour conséquence ses effets délétè-
res (figure 3), et notamment une activation du système
nerveuxsympathiqueaboutissantàuncerclevicieuxneuro-
hormonal.
Dans une sous-étude de Solvd, Francis et al. mon-
traient que l’activité de la rénine plasmatique, et par
conséquent la production d’angiotensine II évoluant
parallèlement, sont ainsi élevés chez les patients insuffi-
sants cardiaques même asymptomatiques mais traités par
diurétiques (bras Prévention, n = 151) et davantage chez
les patients symptomatiques (bras Traitement, n = 80). Un
des objectifs majeurs de la thérapeutique de l’insuffisance
cardiaque est donc de réduire la production d’angioten-
sine II et/ou de bloquer ses effets délétères médiés par les
récepteurs AT
1
.
L’échappement aux IEC
dans le traitement
de l’insuffisance cardiaque
Le traitement par IEC est devenu une pierre angulaire
du traitement de l’insuffisance cardiaque. Les grandes
études cliniques sur l’insuffisance cardiaque des quinze
dernières années (Consensus, Solvd, Save, Trace, Aire...)
ont démontré que l’inhibition du SRA s’accompagnait
d’une diminution de la mortalité, d’une amélioration
symptomatique et de la tolérance à l’exercice, et d’un
ralentissement de la progression de la dilatation ventricu-
laire gauche. Les effets hémodynamiques des IEC sont
résumés dans le tableau 1.
Définition de l’échappement aux IEC
Une réascension progressive plus ou moins rapide des
taux d’angiotensine II peut être observée sous IEC. Ce
phénomène est appelé « échappement aux IEC ».
Avant d’évoquer les différents mécanismes d’échappe-
ment aux IEC, il est nécessaire d’aborder les mécanismes
d’action des IEC ainsi que la réponse du SRA sous IEC.
L’IEC diminue la production d’angiotensine II dépen-
dante de l’ECA et inhibe la dégradation de la bradykinine
ainsi que celle d’autres peptides vasoactifs. Pharmacolo-
giquement, un IEC administré quotidiennement aux poso-
logies recommandées réduit transitoirement les taux
Angiotensinogène
Angiotensine I
Angiotensine II
Récepteurs à
l’angiotensine II
Circulation Tissus
Foie Tissus (cœur, vaisseaux,
rein, cerveau…)
Rénine
rénale
ECA intravasculaire
(endothélium)
Rénine
rénale
ECA tissulaire
(fibroblaste,
muscle lisse)
Fragments inactifs
Fragments inactifs
Ang III, Ang IV
Internalisation
inactivation
Réponses cellulaires
contraction, secrétions
Expression génique
angiotensinogène
ECA, rénine
endothéline...
Systèmes alternes
(chymases…)
Figure 2. Les deux systèmes rénine-angiotensine : tissulaire versus circulant.
mt, vol. 12, n° 2, mars-avril 2006 73
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

d’angiotensine II pendant quelques heures après la prise
orale. Cette réélévation rapide des taux d’angiotensine II,
alors même que l’inhibition de l’ECA persiste, est expli-
quée par la clairance progressive de la molécule et par une
élévation réactionnelle contre-régulatrice de la rénine
plasmatique augmentant l’angiotensine I [9]. Cet excès
d’angiotensine I constitue un substrat pour l’ECA et éven-
tuellement pour les autres systèmes enzymatiques alterna-
tifs au niveau tissulaire (chymase). De plus, l’élévation
réactionnelle de la rénine persiste 24 heures après la prise
médicamenteuse malgré des taux d’angiotensine II reve-
nus à leurs taux initiaux. L’expression de l’enzyme de
conversion augmente également de façon significative en
cas de traitement chronique par IEC.
L’échappement aux IEC n’est pas un problème théori-
que s’exprimant seulement par des données biologiques ;
il a une traduction biologique clinique, pronostique et
échocardiographique.
•Traduction biologique : dans l’étude V-HeFT II
publiée en 1991 [10] randomisant 804 hommes en insuf-
fisance cardiaque, les taux de norépinéphrine diminuaient
davantage sous énalapril que sous l’association
hydralazine-dinitrate d’isosorbide [11]. Cependant, à
24 mois, cette différence s’estompait témoignant d’un
échappement aux IEC.
•Traduction clinique : dans un sous-groupe de l’étude
Consensus I publiée en 1987 [12] enrôlant 253 patients en
Effets délétères de l’angiotensine II
Vasoconstriction artérielle
Élévation de la post-charge
Remodelage vasculaire
Stress oxydant
Hypertrophie des cardiomyocytes
Fibrose myocardique
Apoptose
Activation du système sympathique
Sécrétion d’arginine-vasopressine
Réabsorption sodée directe
Sécrétion d’aldostérone
Atrophie du muscle squelettique
Figure 3. Les effets délétères de l’angiotensine II.
Tableau 1.Effets hémodynamiques de l’inhibition de l’enzyme de
conversion chez les patients en insuffisance cardiaque
Paramètres hémodynamiques Effet
Cardiovasculaires
– Résistance périphérique totale ↓
– Pression artérielle moyenne ↓
– Débit cardiaque et volume d’éjection ↑
– Pré-charge et post-charge ↓
– Pression artérielle pulmonaire ↓
– Fonction diastolique ↑
Rénaux
– Flux sanguin rénal ↑
– Résistance de l’artériole efférente ↓
– Pression intraglomérulaire ↓
– Fraction de filtration glomérulaire ↓
Système nerveux périphérique
– Biosynhèse de norépinéphrine ↓
– Recaptage de l’épinéphrine ↓
– Catécholamines circulantes ↓
Actualités thérapeutiques
mt, vol. 12, n° 2, mars-avril 2006
74
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.

insuffisance cardiaque classe IV NYHA, lorsque les taux
d’angiotensine II diminuaient de plus de 16 pg/mL sous
énalapril, la mortalité à 6 mois était de 7 %, tandis que
lorsque la diminution des taux d’angiotensine II était infé-
rieure à 16 pg/mL, la mortalité à 6 mois était de 21 % [13].
De même, les taux plasmatiques d’angiotensine II mesurés
chez 19 patients sur une période de4à6semaines,
remontaient au-dessus des taux initiaux malgré l’adminis-
tration continue d’énalapril à des doses comprises entre
15 et 20 mg [14]. Dans une série de 70 patients insuffisants
cardiaques traités par IEC [15] à 6 mois, 50 % des patients
avaient des taux élevés d’angiotensine II. Cet échappe-
ment biologique était significativement prédictif d’évène-
ments défavorables (décès, hospitalisation). Hamroff G et
al. [16] montraient que l’addition de losartan à des doses
maximales d’IEC chez des patients insuffisants cardiaques
sévères améliorait leur statut fonctionnel ainsi que leur
consommation d’oxygène au pic d’un effort. Cette amé-
lioration persistante à 6 mois témoignerait également d’un
échappement aux IEC.
•Traduction échocardiographique : les données écho-
cardiographiques de l’étude Save [17] randomisant 2 231
patients après infarctus du myocarde révèlent que, malgré
150 mg de captopril, la progression de la dilatation ven-
triculaire gauche redevient similaire à celle du groupe
placebo après 12 mois de traitement [18]. Cette atténua-
tion des effets antiremodelage des IEC témoigne égale-
ment d’un échappement aux IEC mais aussi de l’histoire
naturelle de la maladie et de la mise en jeu secondaire de
nouveaux systèmes neurohormonaux ou immunitaires de
progression du remodelage ventriculaire gauche.
Les mécanismes responsables de l’échappement aux
IEC dépendent probablement de plusieurs paramètres.
L’administration d’un IEC déclenche elle-même une acti-
vation sur SRA et favorise donc son échappement. Plu-
sieurs solutions peuvent être adoptées :
•Multiplier les administrations. Juillerat et al. [9] mon-
traient effectivement que 5 mg de bénazépril/6 heures
inhibe davantage l’activité de l’ECA qu’une seule prise de
20 mg de bénazépril.
•Utiliser un IEC de demi-vie longue à forte posologie.
Néanmoins alors que l’activité de l’ECA peut être réduite
de 95 %, la diminution de la production d’angiotensine II
ne peut guère dépasser 85 % même au nadir de l’inhibi-
tion de l’ECA c’est-à-dire environ 4 heures après la der-
nière prise [19, 20]. Par conséquent, si l’angiotensine I est
produite en abondance du fait de l’évolution de l’insuffi-
sance cardiaque et de l’élévation réactionnelle de la
rénine sous IEC, les 20 % restants d’angiotensine II repré-
senteront en valeur absolue une production importante.
•Ajouter un antagoniste de l’angiotensine II, ce qui
permettrait de bloquer l’action de l’angiotensine II en aval
de sa production.
Le traitement IEC doit par conséquent être adapté au
niveau de production de l’angiotensine II. Une étude de
Jorde et al. [21] montre que le degré d’inhibition du SRA
(étudié par la réponse tensionnelle à l’injection d’angio-
tensine I) est dose dépendant. Le doublement de la poso-
logie maximale recommandée d’IEC (par exemple passer
de 40 à 80 mg de lisinopril) permet d’inhiber complète-
ment la réponse tensionnelle à l’injection d’angiotensine I
et donc de bloquer efficacement la conversion de l’angio-
tensine I en angiotensine II par l’ECA intravasculaire. En
revanche, la tolérance à long terme de posologies « mas-
sives » d’IEC reste incertaine. D’autre part, doubler la dose
d’IEC est aussi efficace sur la réponse pressive que de
rajouter 80 mg de valsartan, bloquant directement le
récepteur AT
1
à l’angiotensine II [21]. Par ailleurs, la
réduction de la morbidité sous une posologie moyenne de
33 mg de lisinopril en comparaison avec la faible posolo-
gie de 4,5 mg, observée dans l’étude Atlas illustre partiel-
lement les conséquences cliniques de l’échappement aux
faibles doses d’IEC mais aussi les limites des doses maxi-
males recommandées [22].
Les effets des IEC sur le SRA circulant sont relativement
bien étudiés, en revanche l’efficacité des IEC sur la forma-
tion tissulaire d’angiotensine II reste obscure et dépend
probablement de nombreux paramètres comme le type
d’IEC (lipophilie, accessibilité tissulaire de la molécule),
du tissu étudié (rein, cœur, aorte, etc.) et de l’importance
des chymases. Wollert et al. montraient que seule de fortes
doses de lisinopril inhibent l’activité de l’ECA intrarénale
sans affecter celle du ventricule gauche dans un modèle
d’infarctus du myocarde chez le rat [23]. Une autre étude
menée chez le cochon traité par captopril, montrait que le
cœur maintient une production d’angiotensine II, alors
que la production d’angiotensine II circulante est suppri-
mée [24]. Par conséquent, des doses d’IEC ne permettant
pas d’inhiber complètement l’ECA intravasculaire, sont
probablement peu susceptibles d’inhiber l’ECA tissulaire.
Implications thérapeutiques
de l’échappement aux IEC
Les données expérimentales et cliniques démontrent
que l’inhibition du SRA n’est que partielle chez les
patients en insuffisance cardiaque traités avec les doses
maximales recommandées d’IEC. L’inhibition incomplète
de l’ECA entraîne une réascension des taux d’angiotensine
II et une progression symptomatique de l’insuffisance car-
diaque.
Plusieurs options thérapeutiques sont néanmoins offer-
tes :
•L’adjonction de spironolactone a été validée par
l’étude Rales [25]. D’une part, la solution est incomplète
puisque la production d’angiotensine II en amont n’est pas
bloquée et que la stimulation des récepteurs AT1 à des
effets délétères autres que la sécrétion d’aldostérone.
D’autre part, la synthèse d’aldostérone est largement indé-
pendante de la production d’angiotensine II [26] et déter-
minée par de nombreux autres facteurs comme la déplé-
mt, vol. 12, n° 2, mars-avril 2006 75
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 25/05/2017.
 6
7
8
6
7
8
1
/
8
100%