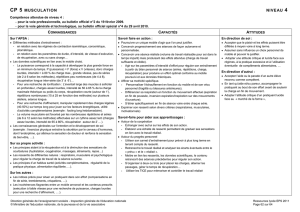
Etude intégrative des répétitions aux niveaux des séquences

Etude intégrative des répétitions
aux niveaux des séquences nucléiques,
des séquences protéiques et
des structures tridimensionnelles des protéines
Atelier de BioInformatique - Université Paris 6
Anne-Laure Abraham

Les duplications intragéniques
Duplication en tandem
Divergence
Nouvelle fonction
sous-fonctionnalisation
Adaptation des organismes
Perte de fonction /
Pathologie
Mutation / sélection
Délétion

Importance de ces duplications
•> 14% des protéines contiennent des répétitions
(Marcotte, 1998)
•Formation de protéines longues
•Permet l’évolution plus rapide des protéines
•Variabilité des protéines en lien avec l’environnement :
Antigènes de surface
Protéines membranaires
Protéines sécrétées
Réponse immunitaire …
Rôle majeur dans l’évolution des protéines

s
sé
équence
quence
nucl
nuclé
éotidique
otidique
s
sé
équence
quence
prot
proté
éique
ique
structure tridimensionnelle des prot
structure tridimensionnelle des proté
éines
ines
?
Comprendre la dynamique
Comprendre la dynamique
de cr
de cré
éation et d
ation et d’é
’évolution des
volution des
r
ré
ép
pé
étitions dans les g
titions dans les gé
énomes
nomes

Plan
•Méthodes pour étudier les répétitions (séquences, structures)
•Données utilisées
•Algorithme de Smith et Waterman / adaptation
•Système de score
•Significativité des résultats (séquences, structures)
•Résultats
•Conclusion - Perspectives
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
1
/
26
100%