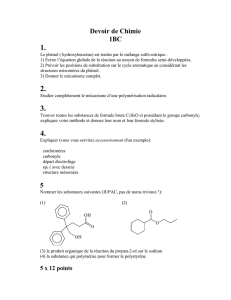
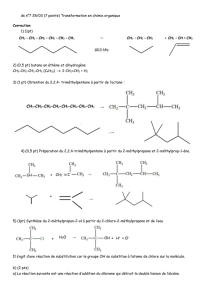
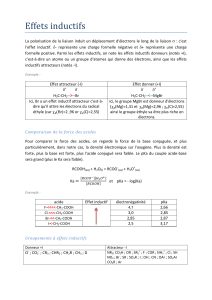
Chap 5 CM8-9 CZ Dong Fichier

1
1 Atome (4h)
2 Décrire une molécule (4h)
3 Interactions intermoléculaires (2h)
4 Introduction à la chimie organique et stéréochimie (5h)
5 Effets électroniques, intermédiaires réactionnels et notions cinétiques (3h)
6 Dérivés halogénés: substitutions nucléophiles – éliminations d’ordre 1 ou 2 (4h)
COURS
De l’atome à la chimie organique
22 heures : 11 séances de 2 heures
Chapitres:

Energie de résonance constatée 2
Cas de butadiène

Energie de résonance constatée 3
Cas de benzène
0

Conjugaison: des doubles liaisons et des simples liaisons sont en conjugaison si elles sont en
alternance. Une double liaison peut être remplacée par une triple liaison, un doublet non liant et
une case quantique (orbitale) vide. Ex: les deux doubles liaisons dans le butadiène.
Dans un système conjugué, la délocalisation des e- p s’étend sur l’ensemble du système.
Ex: les 4 e- p de butadiène sont délocalisés sur les 4 atomes de carbone.
OA p
formation de 4 OM p
dont 2 liantes occupées
(4 e- p venant des 4 OA p)
Energie de résonnance: énergie dégagée grâce à cette délocalisation étendue. Le système est
plus stable car le niveau d’énergie est plus bas.
Mésomérie: Le délocalisation des électrons génère des formes limites de résonance (formes
mésomères). La structure réelle est une superposition de ces différentes formes, certaines ayant
un « poids » plus important (ce sont celles qui correspondent aux énergies les plus basses).
Ex: 3 formes mésomères de butadiène les plus représentatives
1
2
butadiène
4
Conjugaison et Mésomérie

(planéité de la molécule impérative pour la mésomérie)
Cl
NH2
NH2
C O
C O
C O
d+ d-
permet d’interpréter
Cl
Mésomères et représentation d’une molécule
La structure réelle de la molécule correspond à la résonance de toutes formes
mésomères. Pour une question de simplicité, une molécule est toujours
représentée par la forme la plus stable (non chargée). 5
OH OH OH OH OH OH
O
OH
O
OH
O
OH
O
OH
O
OH
O
OH
O
OH
O OH
CH2CH2
CH2
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
1
/
39
100%