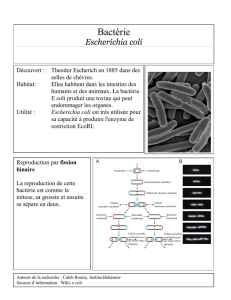
4. Objectif du travail : caractérisation d`une souche Escherichia coli

AVERTISSEMENT
Ce document est le fruit d'un long travail approuvé par le jury de
soutenance et mis à disposition de l'ensemble de la
communauté universitaire élargie.
Il est soumis à la propriété intellectuelle de l'auteur. Ceci
implique une obligation de citation et de référencement lors de
l’utilisation de ce document.
D'autre part, toute contrefaçon, plagiat, reproduction illicite
encourt une poursuite pénale.
Contact : ddoc-theses-contact@univ-lorraine.fr
LIENS
Code de la Propriété Intellectuelle. articles L 122. 4
Code de la Propriété Intellectuelle. articles L 335.2- L 335.10
http://www.cfcopies.com/V2/leg/leg_droi.php
http://www.culture.gouv.fr/culture/infos-pratiques/droits/protection.htm

1
Ecole :
Ecole Nationale Supérieure d’Agronomie et des Industries
Alimentaires
Ecole doctorale :
Sciences et Ingénierie des Ressources Procédés Produits et Environnement
Thèse
présentée pour l’obtention du titre de
Docteur de l’Institut National Polytechnique de Lorraine
en Sciences Agronomiques
par Delphine DUFOUR
Recherche et caractérisation de déterminants génétiques permettant
l’adaptation d’une souche d’Escherichia coli à la mamelle bovine
Soutenue publiquement le 24 octobre 2008 devant la commission d’examen :
Membres du jury :
Rapporteurs : C. Le Bouguènec, Chef de Laboratoire, Institut Pasteur, Paris
C. Burvenich, Professeur, Faculté de Médecine Vétérinaire, Gand, Belgique
Examinateurs : P. Germon, Chargé de recherche, Institut National de la Recherche
Agronomique, Tours
Y. Le Roux, Maître de Conférence, Institut National Polytechnique de Lorraine,
Nancy
A. Dary, Professeur, Université Henri Poincaré, Nancy 1
Unité de Recherche sur l’Animal et les Fonctionnalités des Produits Animaux
Faculté des Sciences – 54506 Vandoeuvre les Nancy

2

3
Remerciements
Merci Messieurs les Professeurs Jean-Luc Gaillard et Guido Rychen de m’avoir accueillie au sein de
votre laboratoire et de la confiance que vous m’avez témoignée pour la réalisation de ce travail.
Merci Madame Annie Dary et Monsieur Yves Le Roux pour tous les savoirs que vous m’avez
transmis, pour le suivi rigoureux et attentif de mes travaux et pour votre gentillesse de tous les
instants.
Merci Madame Chantal Le Bouguènec et Monsieur Christian Burvenich d’avoir accepté d’être
rapporteurs de ce travail.
Merci Monsieur Pierre Germon pour votre accueil lors de mon séjour à Tours, d’avoir accepté de
juger ce travail et surtout pour le suivi, les conseils et l’aide considérable que vous m’avez si
gentiment accordés pour sa réalisation.
Merci Monsieur Gérard Guédon pour vos conseils qui m’ont aidé à mieux comprendre et interpréter
mes résultats.
Merci Emilie pour ta précieuse aide technique de haute qualité et nos bavardages de tout et de rien.
Merci Nawara et Wessam pour votre gentillesse, votre bonne humeur et les moments de franches
rigolades que nous avons eu ensemble. Je vous souhaite une belle réussite pour la suite et d’être
heureux comme vous le désirez.
Merci Monsieur Gérard Humbert de m’avoir accueillie pour la toute première fois au laboratoire et
pour votre gentillesse.
Merci Monsieur Alain Driou pour votre gentillesse.
Merci Madame Catherine Corbier de la confiance que vous me témoignez pour m’avoir accordé des
postes d’ATER à l’IUT.
Merci Chantal, Clarisse, Katy, Madame Lagrange, Laurent, Jean-Michel, Frank, Oukni, Maira,
Aurélie, Emeline, Céline et Faiza.
Papa, je te dédie ce travail, maman, je t’embrasse très fort, et je vous remercie tous les deux d’être
toujours là pour moi et avec moi car j’en ai toujours eu besoin et j’en aurai toujours besoin.
Mon amour et mon petit bébé, je vous aime et vous fais mille bisous tous doux.

4
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
1
/
252
100%