UE 2 Apprentissage des Gestes et Techniques de Base

UE 2
Apprentissage des Gestes et
Techniques de Base
Connaissances nécessaires à la bonne réalisation des travaux
pratiques de chimie
I. La sécurité au laboratoire
II. Le matériel au laboratoire de chimie
III. Techniques courantes en chimie organique
M. Peuchmaur 2011-2012

2
I. La sécurité au laboratoire de chimie
Il existe en salle de Travaux Pratiques certains dangers connus auxquels
le manipulateur se doit d’être sensibilisé. Si celui-ci prend quelques
précautions, le risque d’accident est très faible. Le but de cette
présentation est donc d’apprendre à chacun à modifier son
comportement, afin de mettre hors de danger sa propre personne et son
entourage. Même si, a priori, les manipulations proposées en TP sont
choisies de façon à ne pas présenter de danger, une réaction peut devenir
dangereuse si elle est mal conduite.
I.1. Comportement personnel
- Dès l’entrée dans la salle de TP, le port de la blouse et des lunettes
est obligatoire, même si l’on n’est pas soi-même en train de
manipuler. Le port de lentilles de contact est interdit en TP de
chimie organique, même si l’on utilise en plus des lunettes de
protection.
- Il ne faut jamais manger, boire, fumer ou courir dans une salle de TP.
- Les allées ne doivent pas être encombrées (sacs, vestes… à déposer en
bout de paillasse).
- Les cheveux longs doivent être attachés.
- Porter des vêtements adaptés : pantalon et chaussures fermées.
- Savoir localiser les douches de sécurité et les extincteurs.
- Les gants mis à votre disposition doivent être utilisés
‘intelligemment’.
En cas d’incident, réagir calmement et prévenir un encadrant de TP.
I.2. Produits chimiques
La toxicité des produits chimiques peut être :
- aigüe – une exposition momentanée au produit peut provoquer des troubles graves
chez l’expérimentateur ;
- chronique – les dérèglements physiologiques peuvent apparaître plusieurs mois, voire
plusieurs années après une exposition régulière (exemple : l’amiante).
I.2.1. Savoir se protéger : les gestes élémentaires
Une substance peut entrer en contact avec l’organisme de différentes façons :
- Par ingestion : c’est la voie la plus directe et en général la plus grave. Cependant,
c’est le danger le plus facile à prévenir (ne jamais pipeter à le bouche, ne jamais
consommer d’aliments ou de boissons en salle de TP).
- Par inhalation : cela concerne surtout les gaz et les liquides volatils. Le danger est
double :

3
o Les composés irritants et corrosifs peuvent léser les voies aériennes
supérieures (nez, trachée…) et les poumons (exemple : gaz tels que HCl, Cl2
ou SO2).
o Les composés toxiques atteignent les alvéoles
pulmonaires, passent dans le sang et se répandent dans
l’organisme.
Pour prévenir ce danger, on travaillera sous hotte (=
sorbonne) et on ne tentera jamais de reconnaître un produit
par son odeur.
- Par contact direct avec la peau ou les yeux. Pour prévenir ce
danger, on portera une blouse bien fermé, on utilisera des
gants adaptés lors de la manipulation de produits dangereux,
on portera des lunettes de protection.
I.2.2. Savoir évaluer le danger lié à l’emploi d’un produit
I.2.2.a. Cas des solvants organiques
Leur danger est lié entre autre à la quantité manipulée et à la fréquence
de l’utilisation. Ce risque concerne donc peu la salle de TP où aucun
solvant de toxicité importante n’est utilisé (le benzène est
cancérogène, le CCl4 est responsable de troubles nerveux et
hépatiques…).
En TP, on est essentiellement amené à utiliser de l’éther diéthylique
(éthoxyéthane : CH3CH2OCH2CH3) qui présente un danger de part sa
grande inflammabilité.
I.2.2.b. Cas des réactifs
Au laboratoire, les réactifs sont habituellement utilisés en faible quantité pour minimiser les
risques inhérents à leur emploi. Ces dangers étant mis en évidence sur les étiquettes
commerciales, il est bon de toujours lire ces étiquettes avant tout emploi et de connaître la
signification des différents symboles utilisés.

4
Evolution de l’étiquetage (20-01-2009) :
DANGER DE TOXICITE
AIGUE
DANGERS SUR LA SANTE
(irritant…)
DANGER DE CORROSION
DANGER D’INCENDIE
PRODUITS COMBURANTS
DANGER D’EXPLOSION
DANGERS POUR
L’ENVIRONNEMENT
DANGER POUR LA SANTE
(CMR – Cancérogène, Mutagène
ou toxiques pour la reproduction)
GAZ SOUS PRESSION
I.3. Dangers d’incendie et d’explosion
I.3.1. Le risque d’incendie
C’est un risque important dans tous les laboratoires de chimie,
particulièrement en chimie organique.

5
Principales causes :
- utilisation de solvants volatils et inflammables (éther, alcool, acétone…) ;
- utilisation de réactifs spontanément inflammables et explosifs au contact de l’eau
(exemples : lithium Li, sodium Na, potassium K…) ;
- utilisation de gaz comprimés inflammables (exemple : H2)
Pour prévenir ce danger :
o éviter la présence de source de chaleur au voisinage de vapeurs ou de gaz
inflammables (en particulier ne jamais laisser des solvants à proximité des plaques
chauffantes) ;
o et pour limiter les conséquences, éviter les blouses en matière synthétique.
Pour lutter contre un incendie, les laboratoires disposent
d’extincteurs portatifs dont les plus courants sont :
- les extincteurs à eau pulvérisée pour les feux de classe A :
feux secs (bois, tissus, papiers). Capacité : 6 à 9 litres ;
portée : 2,5 m ; autonomie : 20-30 s.
- les extincteurs à neige carbonique (CO2 solide) pour les
feux de classe B : feux gras (solvants organiques,
hydrocarbures, alcools, huiles, graisses). Capacité : 2, 6 ou
10 kg ; portée : 0,75 à 2,5 m ; autonomie : 20-30 s.
Remarque : ce type de feux peut s’éteindre par
étouffement (couverture, sable).
- les extincteurs à poudre pour les feux de classe B ou C
(feux de gaz). Capacité : 6 ou 9 kg ; portée : 4 à 6 m ;
autonomie : 20-30 s.
Remarque : en cas de feux de gaz, essayer de fermer les vannes pour éviter tout risque
d’explosion.
- les extincteurs à poudre spéciale pour les feux de classe D : feux de métaux (sodium,
magnésium, potassium…).
Sur chaque extincteur est précisé la (ou les) classe(s) de feux sur la(les)quelle(s) il peut être
utilisé.
Remarque : en cas d’incendie, il est souvent possible d’étouffer le feu dans les premières
secondes en obstruant l’orifice du récipient.
I.3.2. Le risque d’explosion
Ce risque est minime en TP (non utilisation de produits explosifs). Attention cependant aux
montages de chimie organique…
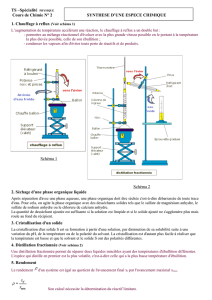
- Ne jamais réaliser de réaction ou de chauffage avec un montage en vase clos (risque
de surpression).
- Ne pas chauffer un liquide sans agitation (agitation manuelle, magnétique ou présence
de grains de pierre ponce).
- Lorsqu’une manipulation est réalisée sous vide (distillation sous pression réduite /
utilisation de l’évaporateur rotatif (rotavapor), séchage sous vide), il y a un risque
d’implosion, avec projection d’éclats de verre lorsque la verrerie utilisée est
défectueuse (ballon étoilé).
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%