TECHNIQUES DE BASE EN CHIMIE ORGANIQUE

!!!!!!!!!!!!!!!!!!!!!!!!!!!!"#$%&!'(!! !!!!!!!!!!!!)&*+,-&!./012./03!
!
0!
!
'456789:4;!<4!=>;4!47!568?84!@AB>789:4!
!
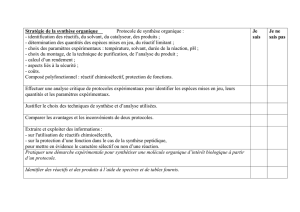
)C!DEFG%+D&!H-ICF#JK&!DK#G!&F!ILFL-CM!M&!D$%LNC!DK#,CFG!O!!
!
!
!
!
!
!
!
!
!
)C!-LC$G#HF!P&!DEFG%+D&!&MM&!NQN&!D&!*C#G!DHK,&FG!PCFD!KF!DHM,CFG!RSHK-!N&GG-&!M&D!-LC$G#*D!&F!$HFGC$GT!&G!U!MVC#P&!PVKF!!"#$%&'(
)'(*+%,--%&'(.(/'-0,1!SHK-!$%CK**&-!M&!N#M#&K!-LC$G#HFF&MW!
!
>! MV#DDK&! P&! !$&GG&! DEFG%+D&X! HF! HYG#&FG! DHK,&FG! KF! NLMCFI&! P&! S-HPK#GW! 8M! &DG! CMH-D! FL$&DDC#-&! P&! N&GG-&! &F! ZK,-&! P&D!
G&$%F#JK&D!S&-N&GGCFG()'(234%/'/('$()5'1$/%6/'!M&!S-HPK#G!PLD#-L!PK!NLMCFI&!-LC$G#HFF&MW!!
!
8M!*CKG! &FDK#G&!DVCDDK-&-!JK&!M&!S-HPK#G!*#FCM! &DG!Y#&F!M&!S-HPK#G!-&$%&-$%LW!8M!*CKP-C! PHF$! $HFFC[G-&! M&D! !3$+")'2( 4'/!'$$%#$(
)56)'#$6-6'/(,#(4/"),6$('$()5'#(*"#$/70'/(0%(4,/'$38(!
!
;#!M&!S-HPK#G!FV&DG!SCD!SK-X!HF!SHK--C!S-H$LP&-!U!KF&!4,/6-6*%$6"#W!
!
:F&!*H#D!M&!S-HPK#G!SK-!HYG&FKX!HF!&F!PLG&-N#F&!MC!JKCFG#GL!P&!NCG#+-&!SHK-!L,CMK&-!M&!-&FP&N&FG!P&!MC!DEFG%+D&W!
! !
! !
;EFG%+D&!
;LSC-CG#HF!
P&D!S-HPK#GD!
5C-C$GL-#DCG#HF!PK!S-HPK#G!
(K-!\!
!
(K-#*#$CG#HF!
@K#!
"#FW!4,CMKCG#HF!PK!
-&FP&N&FG!
7HF!

!!!!!!!!!!!!!!!!!!!!!!!!!!!!"#$%&!'(!! !!!!!!!!!!!!)&*+,-&!./012./03!
!
.!
"8564!0!O!)4!56>:"">B4!>!A4"):]!
!
9,%#)(:!5&GG&!G&$%F#JK&!&DG!KG#M#DL&!SHK-!%**303/'/!RHK!S&-N&GG-&T!KF&!-LC$G#HF!G-HS!M&FG&!U!G&NSL-CGK-&!CNY#CFG&W!!
!
;",/<,"6(:!)C! G&NSL-CGK-&! &DG! KF! *C$G&K-! $#FLG#JK&!O! CKIN&FG&-! MC! G&NSL-CGK-&! S&-N&G! PVC$$LML-&-! MC! -LC$G#HFW! R4F! HKG-&X! M&!
$%CK**CI&!S&-N&G!ILFL-CM&N&FG!PVCKIN&FG&-!MC!DHMKY#M#GL!P&D!-LC$G#*D!PCFD!M&!DHM,CFGWT!7LCFNH#FDX!D#!M&D!-LC$G#*D!HK!M&D!S-HPK#GD!
DHFG! ,HMCG#MDX! #MD! -#DJK&FG! P&! DVL,CSH-&-W! 4F! CPCSGCFG! KF! -L*-#IL-CFG! CK! P&DDKD! PK! DEDG+N&! P&! $%CK**CI&X! M&D! ,CS&K-D! D&!
-&$HFP&FD&FG!&G!-&GHNY&FG!PCFD!M&!YCMMHFW!='(2>2$?!'()'(/'-0,1(4'/!'$()"#*()'(#'(4'/)/'(#6(/3%*$6-2(#6(4/"),6$28(
!
='(!"#$%&'(@(((
!
0!O!AL*-#IL-CFG!U!YHKM&D!
.!O!=CMMHF!
^!O!5%CK**&!YCMMHFX!ou#plaque#chauffante#+#bain#d’huile#(ou#d’eau#si#la#
température#n’excède#pas#100°C)#
1!O!;H-G#&!PV&CK!
3!O!4FG-L&!PV&CK!RSC-!M&!YCD!PK!-L*-#IL-CFGT!
_!O!?LMCFI&!-LC$G#HFF&M!
`!O!;KSSH-G!LML,CG&K-!
!
(
• ='(A%00"#()"6$(B$/'(-613(2"06)'!'#$(.(0%(4"$'#*'(U!MVC#P&!PVKF&!S#F$&!2'//3'(2,/(0%(4%/$6'(/")3'!P&!NCF#+-&!U!G&F#-!DCFD!
MVC#P&!PK!$%CK**&!YCMMHFW!C0()"6$(B$/'(40%*3(2,--62'!'#$(+%,$!SHK-!SHK,H#-!&FM&,&-!-CS#P&N&FG!M&!DEDG+N&!P&!$%CK**CI&!
PK!YCMMHF!&F!YC#DDCFG!M&!DKSSH-G!LML,CG&K-!CK!$CD!Ha!MC!-LC$G#HF!DV&NYCMM&-C#GW!5V&DG!M&!S-&N#&-!LMLN&FG!U!SMC$&-W!
• )&D!-HPCI&D!PH#,&FG!QG-&!I-C#DDLDW!@F!SMC$&-C!SHK-!$&MC!KF! G-C#G!,&-G#$CM!P&!I-C#DD&!DK-!MC!SC-G#&!NbM&!PK!-HPCI&!c!KF&!
-HGCG#HF!P&!MC!,&--&-#&!MH-D!P&!MV&NYH[G&N&FG!S&-N&GG-C!P&!-LSC-G#-!MC!I-C#DD&!KF#*H-NLN&FGW!
• )&!-L*-#IL-CFG!PH#G!&FDK#G&!QG-&!SMC$L!DK-!M&!YCMMHFX!Y#&F!,&-G#$CMX!&G!NC#FG&FK!U!MVC#P&!PVKF&!S#F$&W!D'$$'(46#*'(#'()"6$(
4%2(B$/'(2'//3'X!M&!-L*#-#IL-CFG!&DG!*-CI#M&W!
(
D"#2'602(@((
• >MMKN&-!MC!$#-$KMCG#HF!PV&CK!C,CFG!MC!N#D&!&F!-HKG&!PK!$%CK**CI&!SHK-!,L-#*#&-!MVCYD&F$&!P&!*K#G&DW!
• )H-D!PK! $%CK**CI&! U! -&*MKdX! #M! *CKG! CI#G&-! M&! N#M#&K! -LC$G#HFF&MW! ;#! M&! $%CK**&2YCMMHF! SHDD+P&! KF! P#DSHD#G#*! PVCI#GCG#HF!
NCIFLG#JK&X! HF! SMC$&-C!KF! C#NCFG! RCSS&ML!GK-YKM&FGX! #$#! &F! *H-N&! PVHM#,&T! PCFD! M&! YCMMHFW! ;#! FHFX! HF! PLSHD&! DHK,&FG!
JK&MJK&D!I-C#FD!P&!S#&--&!SHF$&!RHK!P&D!Y#MM&D!P&!,&--&T!SHK-!-LIKM&-!MeLYKMM#G#HFW!!
• :F!G%&-NHN+G-&!Fe&DG!SCD!FL$&DDC#-&!$C-!MC!G&NSL-CGK-&!FeC!SCD!U!QG-&!$HFG-fML&!&F!ILFL-CMW!8M!*CKG!D#NSM&N&FG!$%CK**&-!M&!
DEDG+N&!SHK-!MVCN&F&-!U!LYKMM#G#HFW!!
• 7&!gCNC#D!YHK$%&-!M&!-L*-#IL-CFG!&F!%CKG!O!$&MC!S-H,HJK&-C#G!KF&!LML,CG#HF!P&!S-&DD#HFX!,H#-&!KF&!&dSMHD#HFW!
!
9,'2$6"#2(4/%$6<,'2(@((
• (HK-JKH#!MV&CK!*-H#P&!C--#,&2G2&MM&!SC-!M&!YCD!PCFD!M&!-L*-#IL-CFG!\!!
Ceci#permet#de#remplir#totalement#le#réfrigérant,#et#d’apporter#l’eau#froide#à#l’endroit#où#les#vapeurs#sont#les#plus#chaudes.#
• 9K&!,&KG!P#-&!h!$%CK**&-!CK!-&*MKd!PK!DHM,CFG!S&FPCFG!0/!N#FKG&D!i!\!
Si#dans#un#protocole#apparaît#cette#phrase,#il#faut#compter#les#10#minutes#à#partir#du#moment#où#le#solvant#est#au#reflux,#et#non#
dès#que#le#chauffage#est#mis#en#marche.#
! !

!!!!!!!!!!!!!!!!!!!!!!!!!!!!"#$%&!'(!! !!!!!!!!!!!!)&*+,-&!./012./03!
!
^!
"8564!.!O!)4;!?4'6@<4;!<4!;4(>A>'8@7!
!
EF8!;LSC-CG#HF!PVKF!M#JK#P&!&G!PVKF!DHM#P&!O!MC!GC=HIFHCJK(LJML(NCOP(4'(=5PLLJIFQP!
(
9,%#)(:!>!MC!*#F!HK!CK!$HK-D!PVKF&!DEFG%+D&X!HF!KG#M#D&!$&GG&!NLG%HP&!JKCFP!HF!DHK%C#G&!234%/'/(,#(2"06)'()5,#(06<,6)'W!
(
='(4/6#*64'(@!(HK-!$&MCX!HF!-LCM#D&!KF&!*#MG-CG#HFW!(HK-!JK&!$&MM&2$#!DH#G!SMKD!-CS#P&!&G!SMKD!&**#$C$&X!M&!M#JK#P&!&DG!DHK,&FG!CDS#-L!U!
MVC#P&! PVKF&! G-HNS&! U! &CK! HK! PVKF&! SHNS&! O! HF! SC-M&! CMH-D! P&!*#MG-CG#HF! DHKD! ,#P&! HK! -60$/%$6"#( 2,/( RS*+#'/W! ;#! HF! $%&-$%&! U!
-L$KSL-&-! M&! DHM#P&X! $&GG&! HSL-CG#HF! &DG! CSS&ML&! '22"/%&'( PK! DHM#P&W! ;#! $V&DG! M&! M#JK#P&! JK#! PH#G! QG-&! -L$KSL-LX! HF! SC-M&! P&!
-60$/%$6"#W!
(
='(!"#$%&'(@(
!
0!O!;HM#P&!
.!O!(CS#&-!*#MG-&!
^!O!4FGHFFH#-!=j$%F&-X!!
1!O!"#HM&!U!,#P&!
3!O!"#MG-CG!
_!O!5fF&!&F!$CHKG$%HK$!JK#!CDDK-&!MVLGCF$%L#GL!P&!MC!gHF$G#HF!
(
='(4/"$"*"0'(@(
• )C!*#HM&!U!,#P&!PH#G!QG-&!-613'(%,(AT$6()'(0%(4%600%22'!U!MVC#P&!PVKF&!S#F$&!G-H#D2PH#IGDW!
• U,!6)6-6'/(0'(4%46'/(-60$/'(%V'*(0'(2"0V%#$()'(0%(/3%*$6"#!RHK!M&!DHM,CFG!P&!MC,CI&!M&!$CD!L$%LCFGTW!>gHKG&-!M&!!30%#&'!U!
*#MG-&-!PCFD!M&!=j$%F&-!SK#D!HK,-#-!M&!-HY#F&G!P&!MC!G-HNS&!U!&CKW!;#!HF!$%&-$%&!U!-L$KSL-&-!M&!DHM#P&!R&DDH-CI&TX!-#F$&-!M&!
YCMMHF!C,&$!PK!DHM,CFG!P&!*CkHF!U!Y#&F!-L$KSL-&-!GHKG!M&!DHM#P&W!!
• P*/%2'/(M&!DHM#P&!C,&$(,#($%4"#W! !
• )H-DJK&!GHKG!M&!M#JK#P&!&DG!SCDDL!PCFD!MC!*#HM&!U!,#P&X(*%22'/(0'(V6)'('#()3*"00%#$(0'(RS*+#'/()'(0%(-6"0'(RHK!GHK-FCFG!M&!
-HY#F&G!DK-!M&!GKECK!DV#M!E!&F!C!KFTW(4,62(-'/!'/(0'(/"A6#'$()'(0%($/"!4'(.('%,8(@F!S&KG!&FDK#G&!-L$KSL-&-!M&!DHM#P&!RHK!M&!
M#JK#P&X!D&MHF!MC!S%CD&!JK#!FHKD!#FGL-&DD&TW!
!
D"!403!'#$(@((
D"!!'#$()3A%//%22'/(0'(2"06)'(62"03()'(<,'0<,'2(6!4,/'$32(:!='(0%V%&'(!
(HK-!CNLM#H-&-!MC!DLSC-CG#HF!&G!HYG&F#-!KF!DHM#P&!SMKD!SK-X!HF!S&KG!-LCM#D&-!,#(0%V%&'W!(HK-!$&MCX!HF!KG#M#D&!KF!DHM,CFG!PCFD!M&JK&M!
M&! DHM#P&! FV&DG! SCD! DHMKYM&W! >S-+D! KF! S-&N#&-! &DDH-CI&X! "#( *%22'( 0'( V6)'X! SK#D! "#( %X",$'( 0'( 2"0V%#$( )'( 0%V%&'!RY#&F! *-H#P! P&!
S-L*L-&F$&X!SHK-!N#F#N#D&-!MC!DHMKY#M#GL!PK!S-HPK#GTW!@F!$/6$,/'( 0'(2"06)'!U!MVC#P&!PVKF!CI#GCG&K-!&F!,&--&!RDCFD!I-CGG&-!M&!SCS#&-!
*#MG-&T! SK#D! HF! /'-%6$( 0'( V6)'W! @F! S&KG! -LSLG&-! MVHSL-CG#HF! SMKD#&K-D! *H#DW! 5&GG&! LGCS&! S&-N&G! PVLM#N#F&-! $&-GC#F&D! #NSK-&GLD!
&NS-#DHFFL&D!DCFD!M&!DHM#P&W!
!
• :F&! *H#D! M&! DHM#P&! &DDH-LX! #M! &DG! &F$H-&! %KN#P&!O! PCFD! KF&! $HKS&MM&X! HF! S&KG! M&! SMC$&-! &FG-&! P&Kd! SCS#&-D! *#MG-&! &G! M&!
S-&DD&-!U!MVC#P&!PVKF!GCSHFW!!
!
9,'2$6"#2(4/%$6<,'2(@((
• 5HNN&FG!*C#-&!D#!M&!*#MG-CG!&DG!G-HKYM&!\!
Cela#signifie#que#des#particules#de#solide#sont#passées#à#travers#le#filtre.#Il#faut#alors#recommencer#la#filtration#en#utilisant#un#filtre#
moins#poreux.(#

!!!!!!!!!!!!!!!!!!!!!!!!!!!!"#$%&!'(!! !!!!!!!!!!!!)&*+,-&!./012./03!
!
1!
"8564!.!O!)4;!?4'6@<4;!<4!;4(>A>'8@7!
(
ER8!;LSC-CG#HF!P&!P&Kd!M#JK#P&D!FHF!N#D$#YM&D!O!)>!OPDFKHFHCJK!
(
9,%#)(:!>K!$HK-D!PVKF&!DEFG%+D&X!HF!S&KG!C,H#-!U!234%/'/()',1(4+%2'2(06<,6)'2!FHF!N#D$#YM&DX!&F!ILFL-CM!KF&!S%CD&!H-ICF#JK&!
&G!KF&!S%CD&!CJK&KD&W!
(
='(4/6#*64'(@()&D!P&Kd!S%CD&D!FVCECFG!SCD!MC!NQN&!P&FD#GLX!#M!&DG!SHDD#YM&!P&!M&D!DLSC-&-!SC-!PL$CFGCG#HF!U!MVC#P&!PVKF&!%!4",0'(
.()3*%#$'/W!)&D!S%CD&D!D&!SMC$&FG!P&!YCD!&F!%CKG!SC-!H-P-&!PL$-H#DDCFG!P&!P&FD#GLW!8M!DK**#G!CMH-D!PV#DHM&-!MC!S%CD&!DHK%C#GL&W!
(
!
!
!
='(4/"$"*"0'(@((
(
• lL-#*#&-!JK&!M&!-HY#F&G!&DG!*&-NLW!(MC$&-!KF!YL$%&-!DHKD!MVCNSHKM&!U!PL$CFG&-W!
• ?&GG-&! M&! NLMCFI&! PCFD! MVCNSHKM&W! "&-N&-! MVCNSHKM&!U! MVC#P&! PVKF! YHK$%HFW! A&GHK-F&-!
MVCNSHKM&!&G!SMC$&-! MVHK,&-GK-&!,&-D! M&!*HFP! P&! MC!SC#MMCDD&!RgCNC#D!,&-D! JK&MJKVKF!mTW!@K,-#-! M&!
-HY#F&G!SHK-!)3&%Y'/W!
• A&*&-N&-!M&!-HY#F&G!&G!CI#G&-!,#IHK-&KD&N&FG!MVCNSHKM&W!<LICn&-! P&! FHK,&CK!,&-D!KF!&FP-H#G!
PLICILW!!
• ALSLG&-!MVHSL-CG#HF!GCFG!JK&!PK!ICn!DVL$%CSS&!MH-D!PK!PLICnCI&W!!
• (MC$&-!&FDK#G!MVCNSHKM&!DK-!MVCFF&CKX!'#0'V'/( 0'( A",*+"#!&G!CGG&FP-&!MC!PLN#dG#HF!RDLSC-CG#HF!
P&D!S%CD&DTW!!
• @F!S&KG!&FDK#G&!-L$KSL-&-!MC!S%CD&!,HKMK&!U!MVC#P&!PK!-HY#F&GW!!
(
9,'2$6"#2(4/%$6<,'2(@((
• 5HNN&FG!#P&FG#*#&-!MC!S%CD&!CJK&KD&!&G!MC!S%CD&!H-ICF#JK&!\(
La#phase#la#plus#dense#est#située#en#dessous#:#une#comparaison#des#densités#permet#de#connaître#à#priori#la#position#de#la#phase#
aqueuse.#Néanmoins,#la#présence#de#solutés#peut#changer#la#densité#de#la#phase.#Pour#identifier#avec#certitude#la#phase#aqueuse,#
on#peut#ajouter#quelques#gouttes#d’eau#pour#voir#à#quelle#phase#elles#s’ajoutent,#ou#bien#ajouter#un#volume#conséquent#d’eau#ou#
de#solvant#organique#et#chercher#la#phase#dont#le#volume#augmente.#
!
(
(
D"!403!'#$(O!D"!!'#$(306!6#'/(05'%,(<,6(/'2$'/%6$()%#2(0%(4+%2'("/&%#6<,'(:(='(23*+%&'!
;#!MC!S%CD&!PV#FGL-QG!&DG!MC!S%CD&!H-ICF#JK&X!&G!JKVHF!C!LM#N#FL!MC!S%CD&!CJK&KD&X!HF!S-H$+P&!CS-+D!MC!DLSC-CG#HF!CK!LPDUFQP(OP(
=F( ;UFLP( JIQFKC9MP(C*#F! PVLM#N#F&-! M&D! P&-F#+-&D! G-C$&D! PV&CKW! (HK-! $&! *C#-&X! HF! KG#M#D&! PK! DKM*CG&! P&! NCIFLD#KN! CF%EP-&
MgSO4
W!8M!DVCI#G!PVKF!DHM#P&!G-+D!%EP-HS%#M&W!)H-DJK&!$&GG&!SHKP-&!&DG!,&-DL&!DK-!KF&!S%CD&!H-ICF#JK&!%KN#P&X!&MM&!$CSG&!MV&CK!
&G!DVCIIMHN+-&!O!
MgSO4(s)+ H2O (l) ! →! MgSO4,7H2O
W!
?CF#SKMCG#HF!O!5HNN&F$&-!SC-!N&GG-&! KF&!HK!P&Kd!DSCGKM&D! P&!
€
MgSO4
&G!%&6$'/!U!MVC#P&!PVKF&!YCIK&GG&!P&!,&--&!c!D#! GHKG!M&!
DKM*CG&!P&!NCIFLD#KN!&DG!$HCIKML!CK!*HFPX!&F!CgHKG&-!SK#D!%&6$'/!U!FHK,&CKW!ALSLG&-!MVHSL-CG#HF!gKDJKVU!$&!JK&!M&!DKM*CG&!P&!
NCIFLD#KN!F&!DVCIIMHN+-&!SMKD!&G!*H-N&!KF&!SMK#&!P&!$-#DGCKd!*#F&W!!
>I#G&-!JK&MJK&D!N#FKG&D!U!MVC#P&!PVKF!CI#GCG&K-!NCIFLG#JK&W!!
4M#N#F&-!M&!DHM#P&!SC-!-60$/%$6"#(26!40'!RU!MVC#P&!PVKF!&FGHFFH#-!&G!PVKF!*#MG-&!SM#DDLX!,H#-!$#2$HFG-&TW!
!
!
!
! !

!!!!!!!!!!!!!!!!!!!!!!!!!!!!"#$%&!'(!! !!!!!!!!!!!!)&*+,-&!./012./03!
!
3!
"8564!.!O!)4;!?4'6@<4;!<4!;4(>A>'8@7!
(
ED8!(CDDCI&!PVKF!DHMKGL!PVKF&!S%CD&!U!KF&!CKG-&!O!)VPZHIFDHCJK(4'(=P(=FNFQP!
(
9,%#)(:!4F!*#F!P&!DEFG%+D&X!#M!C--#,&!JKVHF!DHK%C#G&!*C#-&!SCDD&-!PCFD!KF!DHM,CFG!;.!KF!$HNSHDL!>!JK#!D&!G-HK,&!RCK!NH#FD!&F!
SC-G#&T!PCFD!KF!DHM,CFG!;0W!!
o!)H-DJK&!M&!YKG!&DG!P&!-L$KSL-&-!>!PCFD!M&!DHM,CFG! ;.X! HF! P#G! JK&!MVHF!*C#G!KF&! '1$/%*$6"#( P&!>!SC-! M&!
DHM,CFG!;.W!
o!)H-DJK&!M&!YKG!&DG!PVLM#N#F&-!>!P&!MC!S%CD&!;0X!SC-!&d&NSM&!MH-DJK&!$V&DG!KF&!#NSK-&GL!HK!KF!-&DG&!P&!
-LC$G#*X!HF!P#G!JKVHF!S-H$+P&!CK!0%V%&'!P&!;0!SC-!M&!DHM,CFG!;.W!!
!
='(4/6#*64'(@!;0!&G!;.!PH#,&FG!QG-&!FHF!N#D$#YM&DW!4F!ILFL-CMX!MVKF!P&D!P&Kd!&DG!MV&CK!&G!$HFDG#GK&!MC!S%CD&!CJK&KD&X!MVCKG-&!&DG!KF!
DHM,CFG!H-ICF#JK&!RC$LGCG&!PVLG%EM&X!LG%&-X!P#$%MH-HNLG%CF&X!%&dCF&pT!&G!$HFDG#GK&!MC!S%CD&!H-ICF#JK&W!!
@F!KG#M#D&!MC!P#**L-&F$&!P&!DHMKY#M#GLW!F()"6$(B$/'(40,2(2"0,A0'()%#2(0'(2"0V%#$(LE(<,'()%#2(0'(2"0V%#$(L[W!8M!CK-C!CMH-D!G&FPCF$&!U!
JK#GG&-!MC!S%CD&!!;0!SHK-!MC!S%CD&!;.W!
!
='(4/"$"*"0'(@!!
(HK-!M5'1$/%*$6"#!PVKF!S-HPK#G!>!PVKF&!S%CD&!;0!,&-D!KF&!S%CD&!;.!O!HF!CgHKG&!U!MC!S%CD&!U!&dG-C#-&!R;0T!KF!,HMKN&!U!S&K!S-+D!
LJK#,CM&FG!P&!MC!S%CD&!PV&dG-C$G#HF!R;.TW!@F!KG#M#D&!&FDK#G&!KF&!%!4",0'(.()3*%#$'/!R,H#-!.=TX!&G!HF!DLSC-&!M&D!P&Kd!S%CD&D!CS-+D!
C,H#-!Y#&F!CI#GLW!@F!&%/)'(0%(4+%2'()5'1$/%*$6"#(\LE]X!SK#D!HF!/34?$'(05"43/%$6"#( 2,/(0%(4+%2'( .('1$/%6/'(\L[]W!(MKD!HF!-LS+G&!M&!
S-H$&DDKDX!N&#MM&K-!&DG!MV&dG-C$G#HF!RSC-!&d&NSM&X!#M!&DG!SMKD!&**#$C$&!PVKG#M#D&-!^!*H#D!^/!N)!P&!;.!SMKGfG!JKV0!*H#D!q/N)!P&!;.TW!
!
(HK-!M&!0%V%&'!PVKF&!S%CD&!;0!O!HF!CgHKG&!U!MC!S%CD&!U!MC,&-!R;0T!KF!,HMKN&!U!S&K!S-+D!LJK#,CM&FG!P&!MC!S%CD&!P&!MC,CI&!R;.TW!@F!
KG#M#D&!&FDK#G&!KF&!%!4",0'(.()3*%#$'/!R,H#-!.=TX!&G!HF!DLSC-&!M&D!P&Kd!S%CD&D!CS-+D!C,H#-!Y#&F!CI#GLW!@F!&%/)'(0%(4+%2'(0%V3'(
\L[]X!SK#D!HF!/34?$'(05"43/%$6"#(C,&$!KF!FHK,&CK!,HMKN&!P&!S%CD&!P&!MC,CI&!R;.TW!
!
D"!403!'#$(@( D"!!'#$( )6!6#,'/( 05%--6#6$3( )5,#( *"!4"23( "/&%#6<,'( 4",/( 0%( 4+%2'( %<,',2'( \'$( %6#26( )6!6#,'/( 0%( <,%#$6$3(
4'/),'()%#2(0%(4+%2'(%<,',2'](:(='(/'0%/&%&'(
>*#F!)5%!306"/'/(05'1$/%*$6"#()5,#(*"!4"23(V'/2(,#'(4+%2'("/&%#6<,'X!KF!D&M!RG-+D!DHK,&FG!M&!$%MH-K-&!P&!DHP#KN!7C5MRDTT!S&KG!
QG-&!CgHKGL!U!MC!S%CD&!CJK&KD&W!5&GG&!LGCS&X!CSS&ML&!/'0%/&%&'X!S&-N&G!P&!NHY#M#D&-!M&D!NHML$KM&D!PV&CK!&F$H-&!S#LIL&!PCFD!MC!
S%CD&!H-ICF#JK&!RPLD%EP-CGCG#HF!P&!MC!S%CD&!H-ICF#JK&T!&G!P&!P#N#FK&-!MC!DHMKY#M#GL!PK!S-HPK#G!PV#FGL-QG!PCFD!MC!S%CD&!CJK&KD&W!!
4F!&**&GX!M&D!#HFD!7Cr!&G!5M2!HFG!KF&!*H-G&!C**#F#GL!SHK-!MV&CK!O!M&D!NHML$KM&D!PV&CK!DHM,CG&FG!&F!S-#H-#GL!$&D!#HFDX!$&!JK#!&FG-C[F&!
KF&! P#N#FKG#HF! P&! MC! DHMKY#M#GL! P&D! $HNSHDLD! H-ICF#JK&D! PCFD! MV&CK!O! $&D!$HNSHDLD! H-ICF#JK&D! DHFG! -&MC-IKLD! PCFD! MC! S%CD&!
H-ICF#JK&W! )&D! NHML$KM&D! PV&CK! L,&FGK&MM&N&FG! S-LD&FG&D! PCFD! MC! S%CD&! H-ICF#JK&! DHFG! *H-G&N&FG! CGG#-L&D! ,&-D! MC! S%CD&!
CJK&KD&!O!MC!DLSC-CG#HF!P&D!S%CD&D!&DG!CNLM#H-L&W!
! !
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%