Récepteurs AT1 et AT2 de l`angiotensine II : biologie

Act. Méd. Int. - Hypertension (10), n° 8, octobre 1998
200
Au niveau du muscle lisse vasculaire
après activation du récepteur, les
actions intracellulaires de l’angioten-
sine II sont liées à la mise en jeu d’une
phospholipase C dépendant de l’accu-
mulation d’inositol triphosphate qui
provoque la libération de calcium à
partir des stocks intracellulaires, libé-
ration de calcium qui est à l’origine de
la vasoconstriction et, au niveau de la
surrénale, de la libération d’aldostéro-
ne. Parallèlement, il y a stimulation de
la protéine-kinase C qui est à l’origine
des effets à long terme de l’angioten-
sine II : transcription de gènes dans
les cellules musculaires lisses mais
également dans les autres tissus cibles
de l’angiotensine II.
Effets de l’angiotensine II passant
par les récepteurs AT1
Les effets trophiques de l’angiotensi-
ne II ont été obtenus par le groupe du
Pr Dzau sur des cellules myocar-
diques en culture. L’addition de l’an-
giotensine II entraîne une synthèse
d’ARN et de protéines de manière
dose-dépendante. L’angiotensine II,
en stimulant les récepteurs AT1, est un
promoteur de croissance au niveau des
muscles lisses comme au niveau des
cardiomyocytes ou des fibroblastes
myocardiques. Dans les cardiomyo-
cytes, l’augmentation de synthèse de
protéines est associée à une hypertro-
phie de ces cellules. Au niveau des
fibroblastes, l’angiotensine II entraîne
une prolifération cellulaire. Parallè-
lement, l’angiotensine II stimule la
synthèse de collagène. Par ces diffé-
rents effets, la stimulation des récep-
teurs AT1est à l’origine d’un remode-
lage du système cardiovasculaire. Il
convient donc d’opposer deux types
d’action (tableau I) :
- des effets à court terme correspondant à
l’action hémodynamique bien connue de
l’angiotensine II et particulièrement mar-
quée par la vasoconstriction (soit directe
par stimulation du système nerveux sym-
pathique) et à la rétention hydrosodée,
Progrès en hypertension
Récepteurs AT1
et AT2de
l’angiotensine II :
biologie
moléculaire
Michel Andrejak (Amiens)
d'après la communication de V. J. Dzau
(Harvard Medical School, Brigham and
Women's Hospital, Boston, États-Unis)
L’angiotensine II est la
substance biologique-
ment active du système
rénine-angiotensine. Ses
effets sont liés à la stimula-
tion d'un récepteur, le
récepteur AT1, appartenant
à la famille des récepteurs
à 7 domaines transmem-
branaires dont les méca-
nismes d'activation intra-
cellulaire passent par le
couplage à une protéine G
variable selon les tissus.
Les systèmes d'activation
cellulaires sont variés :
adénylate cyclase, phos-
pholipases C, D et A ainsi
qu'un canal calcique.
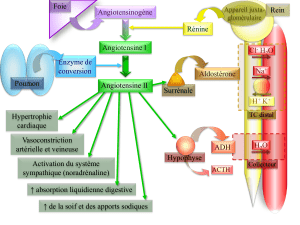
Vasoconstriction :
- directe (effet sur le muscle lisse vasculaire)
- indirecte (actiation sympathique)
Effets à court terme Rétention hydrosodée :
- ➚ réabsorption tubulaire du Na+
- sécrétion d’aldostérone
- vasoconstriction rénale
Sur le myocarde :
- hypertrophie des cardiomyocites
- prolifération des fibroblastes
Effets à long terme - synthèse de collagène
Sur les vaisseaux :
- augmentation de l’épaisseur de la paroi
- prolifération des cellules musculaires lisses
Tableau I : Principaux effets à court terme de l’angiotensine II passant par le biais de récepteurs AT1.

201
- des effets à long terme : hypertrophie
cardiaque, vasculaire, ainsi que des
modifications au niveau du glomérule
(sclérose).
Les effets de l’angiotensine II sur le
myocarde ont été évalués en faisant
appel à une approche transgénique. Il
s’est agi d’utiliser des promoteurs de
chaînes lourdes spécifiques des myo-
cytes du cœur. La surexpression des
récepteurs AT1peut ainsi se faire de
façon spécifique au niveau du myocar-
de sans concerner les autres tissus.
Cette surexpression est létale, avec une
survie inférieure à trois semaines. Le
traitement de ces animaux par le capto-
pril prolonge la durée de survie, ce qui
suggère que l’effet est médié au moins
en partie par l’angiotensine II. La mort
de ces animaux est très brutale et appa-
raît liée à des troubles de conduction.
L’ECG de ces animaux fait état de bra-
dycardie avec allongement de l’espace
PR et élargissement du complexe QRS.
Les récepteurs AT1sont a priori pré-
sents dans le système de conduction et
leur surexpression peut être à l’origine
de troubles de conduction et/ou de
troubles de ryhtme (3).
Chez ces animaux transgéniques les
constatations anatomiques font état de
congestion pulmonaire, d’oreillettes
très dilatées alors que les cavités ven-
triculaires sont seulement dilatées. A la
section du cœur, une hypertrophie des
myocytes et une fibrose apparente de
façon prédominante au niveau des
oreillettes s’expliquent par le fait que le
promoteur pour les chaînes lourdes
s’exprime d’abord dans l’oreillette lors
du développement fœtal. Ce n’est
qu’après la naissance que l’atteinte
concerne également le ventricule. Mais
comme les animaux meurent très
jeunes, les ventricules n’ont pas le
temps de s’hypertrophier. Ces constata-
tions indiquent néanmoins que l’angio-
tensine peut être responsable de remo-
delage cardiaque.
Effets de l’angiotensine II passant
par les récepteurs AT2
Le récepteur AT2de l’angiotensine II,
a été cloné en 1993. D’autres récep-
teurs pourraient également être fonc-
tionnels comme les récepteurs AT4ou
des récepteurs pour l’heptapeptide
(1-7) de l’angiotensine. Il s’agit éga-
lement d’un récepteur à sept
domaines transmembranaires dont la
troisième boucle intracellulaire joue
un rôle important pour les phéno-
mènes de transduction du signal (2).
Sur le plan de la structure, le récep-
teur AT2, bien qu’appartenant à la
même super-famille des récepteurs
liés à la protéine G, est très différent
au niveau des acides aminés constitu-
tifs (32 % seulement sont identiques)
du récepteur AT1.
Le récepteur AT2est surtout exprimé
au cours de la vie fœtale. En fin de ges-
tation, il s’exprime et joue vraisembla-
blement un rôle fonctionnel important.
Par contre, chez l’adulte, on retrouve
peu (ou pas) de récepteurs AT2fonc-
tionnels. Dans certaines circonstances
pathologiques comme l’insuffisance
cardiaque congestive, ce récepteur peut
s’exprimer.
Pour des cellules en culture qui n’ex-
priment normalement que l’AT1et
chez lesquelles on montre que l’an-
giotensine II entraîne une croissance
cellulaire, la mise en jeu supplémen-
taire des récepteurs AT2provoque une
inactivation du rôle promoteur de la
croissance de l’angiotensine II suggé-
rant que les récepteurs AT2peuvent
développer un effet anti-AT1sur les
phénomènes de croissance et de proli-
fération (1).
Le récepteur AT2est couplé à une pro-
téine G qui permet l’activation d’un
système enzymatique tyrosine-phos-
phatase. Cette enzyme tyrosine-phos-
phatase s’oppose indirectement à l’ac-
tion de différentes kinases, en particu-
lier celle de la MAP-kinase (mitogen-
activated protein). La stimulation AT2
aboutit à un effet “anticroissance”.
Dans certaines circonstances spéci-
fiques, la stimulation AT2peut entraî-
ner la mort cellulaire programmée
(apoptose) (5).
Récemment, l’équipe de Dzau a mon-
tré que la stimulation AT2pouvait être
à l’origine d’une interaction avec
d’autres mécanismes impliqués dans
la croissance cellulaire (PDGF, cyto-
kines...). En pratique, la stimulation
de l’AT2paraît inhiber de très nom-
breux facteurs de croissance dans le
système vasculaire. Dzau et son équi-
pe ont suggéré que, pendant la crois-
sance et le développement, ce récep-
teur doit jouer un rôle important de
modulation pour éviter une croissance
excessive en fin de gestation. Chez
l’homme, cette stimulation AT2peut
intervenir dans des circonstances
pathologiques pour inhiber l’action
excessive de la stimulation AT1et des
facteurs de croissance qui intervien-
nent alors.
Cette éventualité a été étudiée in vivo
avec des animaux dépourvus du gène
du récepteur AT2(animaux knock out
ou “KO”). Chez ces animaux, la pres-
sion artérielle de base est légèrement
augmentée mais la réponse pressive à
l’angiotensine II perfusée est très aug-
mentée (majoration de la réponse
vasoconstrictrice). La morphologie
des vaisseaux est très modifiée avec,
au niveau des vaisseaux mésenté-
riques, une augmentation considérable
de la média et du rapport média/lumiè-
re, du fait d’une importante hypertro-
phie vasculaire chez ces animaux où
l’angiotensine ne peut stimuler que
des récepteurs AT1et aucun récepteur
AT2. Chez l’animal normal, au cours
de la gestation, la croissance du vais-
seau est rapide et, en fin de gestation,

Act. Méd. Int. - Hypertension (10), n° 8, octobre 1998
202
Progrès en hypertension
le récepteur AT2est stimulé pour inter-
rompre la croissance et permettre la
différenciation (remodelage vasculaire
de fin de vie fœtale). Chez les animaux
“KO”, il y a excès de stimulation AT1
et des facteurs de croissance, sans que
cette stimulation ne puisse être antago-
nisée par la mise en jeu de récepteurs
AT2. Les vaisseaux sont plus épais et
les animaux apparaissent prédisposés à
la vasoconstriction et au développe-
ment de l’hypertension artérielle.
Si l’on provoque une hyperplasie de l’in-
tima par constriction de l’artère fémora-
le de l’animal au moyen d’un collet en
plastique, une réexpression locale du
récepteur AT2intervient pour limiter les
phénomènes de croissance. Chez les ani-
maux “KO” pour le récepteur AT2, il n’y
a pas de réexpression des récepteurs
AT2, et les lésions de l’intima au cours
de la procédure d’agression de la paroi
artérielle sont beaucoup plus impor-
tantes. C’est un argument supplémentai-
re pour indiquer que les récepteurs AT2
se réexpriment dans les situations où il y
a surexpression du récepteur AT1.
Au niveau du myocarde, beaucoup de
travaux ont été consacrés aux consé-
quences de l’expression des récepteurs
AT1et AT2. Dans les conditions expéri-
mentales où peut être produite une
hypertrophie du myocarde (infarctus
expérimental), le rapport AT2/AT1aug-
mente par réexpression des récepteurs
AT2visant à moduler les effets AT1.
Dans un modèle d’insuffisance car-
diaque, par ligature des coronaires (Liu
et coll.), une hypertrophie cardiaque,
avec augmentation de taille des myo-
cytes et excès de collagène dans l’in-
terstitium myocardique, se développe.
Le traitement par AT1-bloquant pré-
vient ces phénomènes. Par contre, l’as-
sociation anti-AT1et anti-AT2suppri-
me les effets favorables exercés par les
AT1administrés seuls. Cela suggère
qu’une partie des effets favorables des
AT1-bloquants passent par une stimula-
tion AT2dont est responsable l’angio-
tensine II qui circule en grande quanti-
té du fait du blocage des récepteurs AT1
(4).
Le tableau II indique les conséquences
de la stimulation respective des récep-
teurs AT1et AT2.
Références
1) Dzau V.J., Horiuchi M. : Differential
expresssion of angiotensin receptor sub-
types in the myocardium : a hypothesis.
Eur. Heart J., 1996, 17 : 978-980.
2) Hayashida W., Horiuchi M., Dzau V.J. :
Intracellular third loop domain of angio-
tensin II type-2 receptor. Role in mediating
signal transduction and cellular function.
Journal Biol.Chem., 1996, 271 (36) :
21985-21992.
3) Hein L., Stevens M.E., Barsh G., Pratt
R.E., Kobilka B.K., Dzau V.J. : Overexpres-
sion of angiotensin AT1receptor transgene
in the mouse myocardium produces a lethal
phenotype associated with myocyte hyper-
plasia and heart block. Proc. Natl. Acad.
Sci. USA, 1997, 94 : 6391-6396.
4) Liu Y.H., Yang X.P., Sharov V., Nass O.,
Sabbah H., Peterson E., Carretero O.A.
Effects of angiotensin-converting enzyme
inhibitors and angiotensin II type 1 recep-
tor antagonists in rats with heart failure.
Role of kinins and angiotensin II type 2
receptors. J. Clin. Invest., 1997, 99 : 1926-
1935.
5) Yamada T., Horiuchi M., Dzau V.J.
Angiotensin II type 2 receptor mediates
programmed cell death. Proc.Natl. Acad.
Sci. USA, 1996, 93 : 156-160.
Propos recueillis par le Pr Michel Andrejak au cours des journées de l’HTA, décembre 1997.
Tableau II. Mise en jeu des récepteurs AT1et AT2de l’angiotensine II.
AT1AT2
vasoconstriction vasodilatation
Conséquences de la croissance cellulaire inhibition de la croissance cellulaire
stimulation prolifération différenciation cellulaire
antinatriurèse apoptose
radicaux libres natriurèse
sécrétion d’aldostérone production de monoxyde d’azote (NO)
libération d’endothéline,
de catécholamines, molécules
d’adhésion, facteurs de croissance
Expression permanente réexpression seulement dans des situations
pathologiques
1
/
3
100%