Thermodynamique (partie II) Fluides réels et transitions de

UNIVERSITE Paris-Diderot
Préparation à l’Agrégation Interne de Physique 2013-14
Séance du 18 Décembre 2013 - M. Mouchet (martine.mouc[email protected])
Thermodynamique (partie II)
Fluides réels et transitions de phases
Notes de cours et illustrations en annexe
Thème 1 - Fluides réels
I - Equation d’état du gaz de van der Waals (1873)
1. Expression
2. Description microscopique : interprétation des coefficients a et b
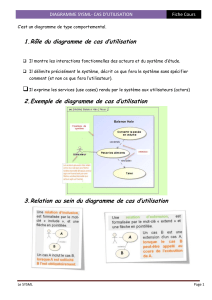
II - Propriétés physiques
1. isothermes d’Andrews p(V)
2. paramètres critiques
3. variables réduites : principe des états correspondants
III - Autres équations d’état
IV - Application aux détentes
1. détente de Joule Gay-Lussac
2. détente de Joule Thomson
(a) température d’inversion
(b) application à la liquéfaction des gaz
Thème 2 - Transitions de phase d’un corps pur
I - Exemples illustratifs et propriétés remarquables
II - Transitions solide/liquide/gaz : diagrammes de phase ; notions de points triple et critique
1. Expérience de base
2. Diagrammes généraux de phase
(a) diagramme (P,V) : isothermes d’Andrews
(b) diagramme (P,T) : point triple et point critique
(c) théorème des moments
III - Relation de Clapeyron
1. Etude thermodynamique
2. Chaleur latente de changement d’état
3. Bilan thermique pour une transformation réversible élémentaire sur le palier d’équilibre
4. Etablissement de la relation de Clapeyron
(a) formulation
(b) expression approchée pour une transformation L−→ G
(c) cas de la courbe de fusion
5. Caractéristiques du point critique C
(a) fluide hypercritique
(b) expérience des tubes de Natterer
IV - Notion succincte d’états métastables - Lien avec l’équation de van der Waals des fluides
1. Exemples

2. Où l’on retrouve les fluides réels
(a) régions instables et métastables du diagramme (P,V)
(b) construction de Maxwell
(c) courbe spinodale
V - Notion d’ébullition
VI - Transitions d’ordre supérieur
1. Classification d’Ehrenfest
2. Classification de Landau : paramètre d’ordre et brisure de symétrie
Références bibliographiques complémentaires
–Atkins, P.W., de Paula, J., “Chimie physique”,DeBoeck(voiraussiversionWEBinteractive
http ://www.whfreeman.com/pchem7/ )
–Bruhat, G., “Thermodynamique”,Masson
– Callen, H.B., “Thermodynamics and an introduction to thermostatistics”,JohnWileysandSons
–Lavertu, G., “Thermodynamique”,Vuibertsupérieur,SériePlatine
–Papon, P., Leblond, J., Meijer, P.H.E., “ Physique des Transitions de phases”,Dunod(niveau
Masters permettant d’aborder des changements d’états hors S-L-G)
–Renvoizé, V., “Thermodynamique : fiches, méthodes et exercices corrigés”,Ellipses
–Tabor, D., “Gases, liquids and solids”, Cambridge University Press
– Woods, L.C., “The thermodynamics of fluid systems”, Clarendon Press
Sur le Web :
Illustrations de diagrammes de phase :
– http ://dao.mit.edu/8.231/PurePhases.htm (A Menagerie of Pure Component Phase Diagrams)
– http ://www.physics.ohio-state.edu/∼wilkins/group/phases/index.html
Animations :
– http ://www.youtube.com/watch ?v=cSli089x7UU (visualisation du passage par l’opalescence cri-
tique)
– http ://www.univ-paris-diderot.fr/Mediatheque/spip.php ?article237 (première mondiale (Oct. 2011)
de la fabrication d’un mag-surf à Paris-Diderot)
Données thermodynamiques de fluides :
–http ://webbook.nist.gov/chemistry/fluid/
– http ://encyclopedia.airliquide.com/encyclopedia.asp ?LanguageID=2&GasID=26
Comment faire pratiquement pour connaître l’état d’un fluide ?
a) Soit on connait Pet T:
-siP>Ps(T):c’estunliquide
-siP<Ps(T):c’estungaz
-siP=Ps(T):lesdeuxphasesco-existent.Lafractionxgde gaz est donnée par la mesure du volume
(molaire).
b) Soit on connait Tet V:
On suppose d’abord que le fluide est gazeux et on applique l’équation d’état du gaz (supposé parfait
si le gaz est dilué). Si la pression trouvée est inférieure à Ps(T),lefluideesteffectivementgazeux.Sion
trouve P>P
s(T),lefluidepeutêtresoitentièrementliquide,soitàl’équilibreliquide-gaz.Pourtrancher,
il faut connaitre l’équation d’état du liquide.
On peut aussi, si on connait le diagramme (P,V) du fluide, regarder dans quelle partie du diagramme
est le point représentatif de l’état du système :
-àl’intérieurdelacourbedesaturation:onaco-existenceduliquideetdugaz
-àdroite:c’estungaz
-àgauche:c’estunliquide.



 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
1
/
31
100%