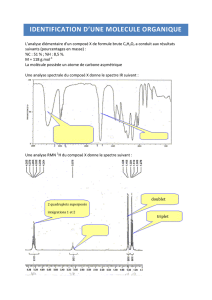
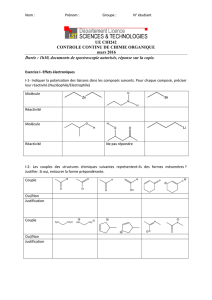
Cours de Spectroscopie moléculaire 1ere année MASTER en

1
Université d’El-Oued
Faculté des Sciences Exactes
Cours de Spectroscopie moléculaire
1ere année MASTER en
Physique appliquée : rayonnement et énergie
2015-2016
Dr. ZEROUAL Soria
Maître de conférences B

2

3
Programme
I- Généralités ………………………………………………………………………...…….. 4
II- Symétrie moléculaire et théorie des groupes ………………………………………...…… 12
III- Absorption IR et diffusion Raman ………………………………………………..……. 26
IV- Spectres électroniques des molécules ……………………………………………………... 42
V- Résonance magnétique nucléaire ……………………………….…………………….. 53

4
Chapitre I:
Généralités

5
I-1- Définition de la spectroscopie moléculaire:
La spectroscopie moléculaire ( SM) consiste à faire passer un rayonnement électromagnétique de
différentes fréquences à travers un échantillon d'un composé et à déterminer le mode d'absorption
de ce rayonnement.
Le spectre résultant donne l'énergie absorbée ou transmise en fonction de la fréquence du
rayonnement.
La SM est très utile et ses applications sont nombreuses: identification de composés, dosage,
imagerie, analyse physique; et elle s'adapte à tous les types d'échantillons.
Elle présente un bon outil pour vérifier les calculs de la mécanique quantique. Si les valeurs
spectrales sont en bon accord avec les différences entre les niveaux d'énergie calculés par la MQ,
cela indique que les calculs sont bon.
I-2-Energies moléculaires et régions du spectre électromagnétique:
Une molécule isolée possède plusieurs types d'énergie:
1- Energie de translation: cette énergie est due au mouvement de la molécule dans son
ensemble, elle n'est pas quantifiée, et ne donne pas lieu à une spectroscopie (E 200cm-1 à
température ambiante).
2- Energie rotationnelle: due à la rotation d'une molécule autour d'un axe passant par son centre
de gravité, elle correspond au domaine d'énergie des micro-ondes ( allant de 1-100cm-1).
Toutes les molécules ayant un moment dipolaire permanant sont active en micro-ondes.
3- Energie vibrationnelle: due au déplacement périodique de ses atomes de leurs positions
d'équilibre; elle correspond au domaine des rayonnement IR (200cm-1 – 4000cm-1).
4- Energie électronique: due au mouvement des électrons de chaque atome, qui correspond au
domaine UV-Visible.
5- Energie magnétique nucléaire: due à l'interaction des noyaux placés dans un champ
magnétique intense; c'est le domaine de la RMN.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
1
/
60
100%