AAC inflammatoire

Les manifestations inflammatoires et
vasculitiques de l’angiopathie
lïd ééb l
amy
l
o
ïd
e c
é
r
éb
ra
l
e
Alexandre C Samak1, Manon Bélair1,
France Berthelet2, Sylvain Lanthier3
1: Département de radiologie, Centre Hospitalier de l’Université de Montréal
2: Département de pathologie, Centre Hospitalier de l’Université de Montréal
3: Département de neurologie, Centre Hospitalier de l’Université de Montréal

Objectifs
•Connaître les manifestations radiologiques et pathologiques
classiques de l'angiopathie amyloïde cérébrale.
•Savoir qu'il existe des manifestations inflammatoires et
vasculitiques rares de l'angiopathie amyloïde cérébrale.
•Connaître les manifestations radiologiques des formes
inflammatoires et vasculitiques de la maladie.
•Connaître les trouvailles pathologiques associées à ces deux
formes.
•Connaître l'évolution naturelle et sous traitement de cette
maladie.

Plan
•Introduction
•Angiopathie amyloïde cérébrale inflammatoire
–Caractéristiques cliniques
–Caractéristiques pathologiques
–Caractéristiques radiologiques
•Présentation de cas
•Angiopathie amyloïde cérébrale inflammatoire
–Traitement et pronostic
–Critères diagnostics
•Résumé

Introduction
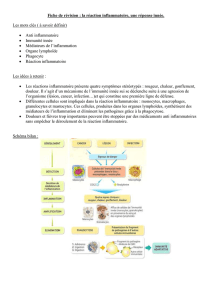
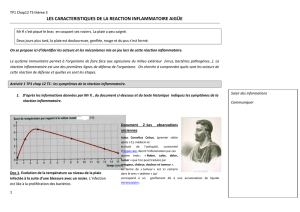
1. L’angiopathie amyloïde cérébrale (AAC):
–Caractérisée par des dépôts de la protéine béta-amyloïde dans la
média et l'adventice des artères corticales et leptoméningées de
petit et moyen calibre
–Démontrée chez 27-32% des personnes âgées asymptomatiques
2. L’imagerie médicale joue un rôle critique dans le
diagnostic et le suivi de l’AAC classique et
inflammatoire
Eisiri and Wilcock, 1986

Introduction
•L’AAC entraîne
–une nécrose fibrinoïde des vaisseaux,
–la formation de microanévrysmes,
–un rétrécissement luminal et de l’ischémie de la
è
mati
è
re blanche
•Présentations cliniques les plus fréquentes sont:
–Les hémorragies lobaires
–Le déclin cognitif
–Les accidents ischémiques transitoires, souvent
répétés et stéréotypés
Greenberg et al, 1993
Maia et al, 2007
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
1
/
128
100%