c. La détermination du moment dipolaire

CHAPITRE 5
L’état liquide (2)
Préambule
Le chapitre précédent n’a pas épuisé l’étude des propriétés observables associées à l’état
liquide. Ce chapitre n’est donc en quelque sorte que la continuation du précédent. On y
présente les propriétés optiques et électriques.
1. Les propriétés optiques
1.1. La réfractométrie
a. Définition
Un rayon de lumière, dans un milieu homogène se propage en ligne droite. Si le milieu
traverse des milieux non homogènes le déplacement de ce faisceau lumineux ne se fait plus en
ligne droite. On aura déjà «vu» ce phénomène au moins de manière indirecte. Ainsi le
pécheur au bord de l’eau voit la ligne de canne à pêche qui ne suit pas une ligne droite : la
ligne fait un angle différent de 180 à la surface de l’eau. Il en est de même d’un faisceau
lumineux qui passe de la phase air à l’intérieur de l’eau : du milieu m au milieu M de la figure
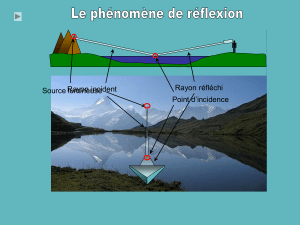
5.1. La formation des «mirages» dans le désert s’explique par ce phénomène
Figure 5.1. Description du principe de la réfraction.

Au moment où le faisceau lumineux passe d’un milieu m à un autre M, la loi qui gouverne ce
comportement est la suivante : le rapport des vitesses v d’un faisceau de lumière dans deux
milieux différents m et M est égal au rapport des sinus de l’angle d’incidence à l’angle de
réfraction. Sur la figure 5.1, on obtient :
5.1
Il faut rappeler que si la vitesse de la lumière est c dans le vide, alors c =
où est la
fréquence et
la longueur d’onde de la lumière.
En pratique le milieu m est l’air et n est une caractéristique qui dépend donc du milieu par
rapport à l’air. En valeur absolue, la valeur de n dans le vide est égale à l’unité. Dans l’air, la
même valeur est très proche de celle de l’air. La valeur de n de l’équation 5.1 n’est autre que
l’indice de réfraction du milieu.
On voit aussi que lorsque le faisceau lumineux arrive de manière rasante, rayon DO, sur la
surface, l’angle de réfraction atteint une valeur maximum, rmax (figure 5.2). Ce rayon ressort
selon OE. En vertu du principe de retour inverse du chemin de la lumière, si le faisceau de
lumière passe d’un milieu dans lequel l’indice de réfraction
M est supérieur à celui
m du
second milieu, seuls les rayons dont l’angle d’incidence est inférieur à rmax de la figure 5.1,
seront réfractés dans le milieu d’indice
m. Le rayon lumineux BO ressort selon OA. Si les
rayons incidents ont un angle d’incidence supérieur à rmax ils seront réfléchis sur la surface du
milieu, surface qui agira alors comme un miroir : rayon MO par exemple.
Figure 5.2. Réfraction maximum. Lorsque le rayonnement passe d’un milieu d’indice de
réfraction supérieur vers un milieu d’indice de réfraction supérieur :
m <
M .
b. La réfraction

La valeur de n dépend de plusieurs facteurs. On peut en identifier plusieurs. On écrit ainsi
que n est une fonction de la température, de la longueur d’onde de la lumière considérée, de la
concentration du soluté (ou des solutés) lorsque l’on parle de solution, de la pression, …
n = f(T,
, [CM], P, …)
Définissons la réfraction spécifique R :
5.2
Cette grandeur R est indépendante de la température et de la pression. Selon la théorie
électromagnétique, on montre que la réfraction molaire RM est telle que :
5.3
Dans cette formule M est la masse molaire de la molécule. Cette relation a pour conséquence
que l’on peut décomposer la réfraction molaire d’une molécule en ses composantes
atomiques. En effet, la masse de la molécule est égale à somme des masses Mi des atomes
constitutifs ;
M = i Mi
Par conséquent,
RM = i RMi
On peut donc établir un tableau donnant les valeurs des réfractions molaires par atome ou
groupe d’atomes. Ce tableau relativement court permet de prévoir et de calculer les
réfractions molaires de n’importe quelle molécule. Le tableau 5.1 donne les valeurs obtenues
des réfractions molaires par atome ou groupes d’atomes.
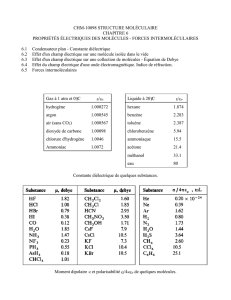
Tableau 5.1. Valeurs des réfractions molaires par groupe d’atomes.
Groupe
d’atomes
nD
Groupe
d’atomes
nD
Groupe
d’atomes
nD
Cl
5,844
O (éthers)
1,643
S (sulfures)
7,729
Br
8,741
O (acétates)
1,61
=S
7,921
I
13,954
OH (alcools)
2,55
NH2 (amines)
4,44
F
0,81
SH (thiols)
8,76
NH (amines)
3,61

H (dans CH2)
1,028
CO (acétone)
4,60
NO
6,71
C (dans CH2)
2,591
CO (méthyl-
cétones)
4,76
ONO (nitrites)
7,24
CH2
4,65
COO (esters)
6,20
NO (nitroso)
5,2
CH3
5,65
COOH
7,23
CO3
7,7
C=C
6,757
CC
7.159
SO3
11,34
Cycle à 3
maillons
0,614
CN
6,46
PO4
10,77
Cycle à 4
maillons
0,317
SCN
13,40
N=C=S
15,62
n-C4H9
19,59
C3H5 (allyle)
14,52
Nota : raie D du sodium ;
nD est en cm3/mol
ou m3/mol 106 à 20 C
iso-C4H9
19,62
C6H5 (phényle)
25,463
tert-C4H9
19,85
C10H7
(naphthyle)
43,00
Ce tableau montre que le groupe CH2 a une réfraction molaire qui est bien égal à la somme
des réfractions atomiques de 2 atomes d’hydrogène ajoutée à celle d’un atome de carbone.
On voit aussi la structure des entités moléculaires n’est pas sans effet. Ainsi, les réfractions
molaires des 3 radicaux C4H9 sont légèrement différentes d’un radical à l’autre et différentes
de la somme 4 RM(C) + 9 RM (H) = 19,63. Ces écarts sont encore plus importants lorsque des
modifications structurales sont importantes comme dans le radical allyle C3H5 ou le radical
phényle C6H5.
Au lieu de constituer le tableau des réfractions molaires par atome ou groupe d’atomes, on
peut aussi le faire selon la nature des liaisons présentent dans les molécules. C’est ce que
montre le tableau 5.2. Dans ce cas, la réfraction molaire est une propriété additive des
liaisons.
Tableau 5.2. Valeurs des réfractions molaires des liaisons.
Liaisons
Incrément
Liaisons
Incrément
Liaisons
Incrément
CH
1,676
CC dans un cycle
C3
1,49
NO
2,43
CC
1,296
CC dans un cycle
C4
1,37
N=O
4,0
C=C
4,17
CC dans un cycle
C5
1,26
N=N
4,12
CF
1,55
CC dans un cycle
C6
1,27
CS
4,61

CCl
6,51
C=O (ethers)
1,54
C=S
11,91
CBr
9,39
CO (acétates)
1,46
CN
1,57
CI
14,61
C=O (cétones)
3,32
C=N
3,76
C=O (méthyl
cétone)
3,49
CN
4,82
OH (alcohols)
1,66
OH (acides)
1,80
Nota : raies D du sodium ; incrément en cm3/mol ou m3/mol 106 à 20 C.
Les valeurs sont bien entendues différentes de celles présentes dans le tableau 5.1. En plus
des valeurs par liaison, on doit tenir compte encore des effets de structures. Par exemple dans
le cas du cyclopropane, un cycle à trois maillons est très tendu. Il implique une modification
des orbitales électroniques : on verra plus tard le lien qui existe entre la réfraction molaire et
le nuage électronique (voir le cours Chimie théorique, chapitre 13.10). Il y a lieu d’introduire
des corrections, des incréments, qui tiennent compte de ces facteurs structuraux.
En résumé, hormis ces corrections d’ordre structural, on dit que la réfraction molaire est une
propriété additive des atomes (tableau 5.1) ou des liaisons (tableau 5.2) constitutifs de la
molécule.
c. La mesure de l’indice de réfraction
Le réfractomètre est un appareil relativement simple : c’est peut-être le premier appareil de
mesure après le thermomètre que l’étudiant a eu l’occasion d’utiliser. Il est essentiellement
formé d’une lampe à vapeur de sodium qui a la caractéristique d’émettre un spectre de
lumière presque monochromatique (au moins à cause des intensités des raies émises). En fait
le sodium émet plus de 99 % de la lumière sous forme d’un doublet situé dans le jaune : 589,0
et 589,6 nm. Ce sont les raies D du sodium, d’où l’appellation nD pour l’indice de réfraction.
La différence de longueur d’onde étant si petite, que pour la réfractométrie, on peut considérer
le faisceau comme monochromatique. Ce faisceau de lumière passe à travers le liquide dont
on veut mesurer l’indice de réfraction. L’appareil est directement calibré ce qui permet une
lecture directe de la mesure.
Une quantité de liquide inférieure à 1 mL suffit. On obtient un résultat compris le plus
souvent entre 1,33 et 1,75 avec au moins 5 chiffres significatifs. La sensibilité est 0,000 05
dans le cas du réfractomètre de PULFRICH et de 0,0002 avec le réfractomètre de ABBE (T
étant inférieur à 0,2 ºC). Ce dernier appareil est très fréquent dans les laboratoires.
Un détail complémentaire, l’appareil est thermostaté pour permettre d’éviter des variations de
l’indice de réfraction dues à la variation de température. En général cette température est
maintenue à 20,0 C ± 0,1 C, sauf si le composé dont on veut connaître l’indice est solide à
cette température.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
1
/
33
100%