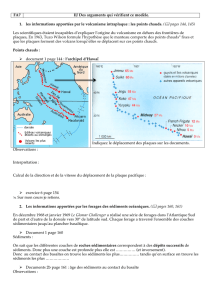
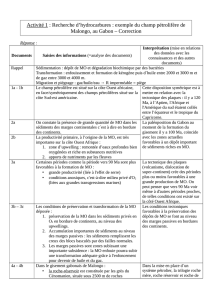
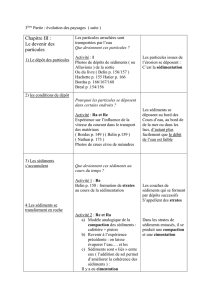
étude sur la biodisponibilité des métaux dans les sédiments

www.eau-artois-picardie.fr
ÉVALUATION DE LA BIODISPONIBILITÉ
DES MÉTAUX DANS LES SÉDIMENTS
2010


(YDOXDWLRQGHODELRGLVSRQLELOLWpGHVPpWDX[GDQV
OHVVpGLPHQWV
3KDVH,,²5DSSRUWILQDO
Rapport Scientifique
G. Billon, R. Descamps, C. Grare, B. Ouddane et J-C. Fischer
Juillet 2010
Gabriel Billon
USTL/Géosystèmes/ECAM
Cité Scientifique, Bât C8, 2
ème
étage
gabriel.billon@univ-lille1.fr
Tel : +33 (0)3 20 43 49 28
Fax : +33 (0)3 20 43 48 22
Gé systèmes
FRE 3298 CNRS-LILLE1

ii

iii
REMERCIEMENTS
Nous tenons à remercier tout d’abord M. Jean Prygiel, chef de service Ecologie du Milieu de l’Agence
de l’Eau Artois-Picardie pour l’aide apportée tout au long de ce travail réalisé dans le cadre de la
convention de coopération USTL-Agence de l’Eau (convention de coopération 64556).
Un grand merci également à Michel Arold de l’Agence de l’Eau Artois-Picardie pour son aide sans
faille lors des prélèvements sur l’ensemble du territoire de la région Nord Pas-de-Calais.
Enfin, je tiens à remercier très chaleureusement M. Yue Zhu, étudiant en Master II dans notre équipe,
ainsi que M. Pierre-Jean Superville, étudiant à l’Ecole Nationale Supérieure de Chimie de Lille, pour
les centaines d’analyses effectuées sur les sédiments et les eaux interstitielles et surnageantes.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
1
/
42
100%