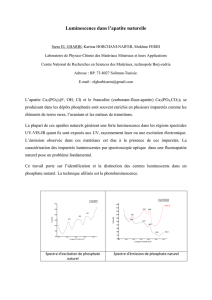
Thèse Sanaa Saoiabi

UNIVERSITÉ MOHAMMED V – AGDAL
FACULTÉ DES SCIENCES
RABAT
N° d’ordre : 2547
THÈSE DE DOCTORAT
Présentée par
Sanaâ SAOIABI
Discipline : Chimie
Spécialité : Matériaux et Environnement
Matériaux fonctionnels à base de phosphate de calcium
à applications environnementales
Soutenue le 28 Octobre 2011
Devant le jury :
Président :
A. ZRINEH, Professeur à la Faculté des Sciences, Rabat Maroc
Examinateurs :
T. CORADIN, Directeur de recherche, CNRS- Paris, France,
M. HAMAD, Professeur à la Faculté des Sciences, Rabat, Maroc
J. NAJA, Professeur et Vice Doyen de la Faculté des Sciences et Technique, Settat Maroc
J. L. ACKERMAN, Professeur à Harvard Medical School, Boston, MA USA
P. BARBOUX, Professeur à l’Ecole Nationale Supérieure de Chimie de Paris, France
A. LAGHZIZIL, Professeur à la Faculté des Sciences, Rabat- Maroc
Faculté des Sciences, 4 Avenue Ibn Battouta B.P. 1014 RP, Rabat – Maroc
Tel +212 (0) 5 37 77 18 34/35/38, Fax : +212 (0) 5 37 77 42 61, http://www.fsr.ac.ma

AVANT PROPOS
Les travaux présentés dans cette thèse ont été réalisés au sein du Laboratoire de
Chimie Physique Générale à la Faculté des Sciences de Rabat (FSR) - Université Mohammed
V-Agdal–Rabat, sous la direction du Professeur Abdelaziz LAGHZIZIL, qui a agréablement
assuré l’encadrement de cette thèse avec beaucoup d'intérêt et d'optimisme tout en me laissant
une large marge de liberté pour mener à bien ce travail. Je tiens à lui exprimer ma gratitude
pour son suivi, sa disponibilité, sa compréhension, son respect des engagements et pour ses
encouragements continus. Je le remercie aussi pour sa patience et son sérieux dont il a fait
preuve lors des multiples discussions et pour l’intérêt constant qu’il a manifesté pour mes
recherches et ses conseils éclairés au cours du développement de ce travail. Sa passion pour la
recherche, son esprit innovateur et son dynamisme perpétuel sont exemplaires. Etre son
étudiante fut un grand honneur pour moi.
Ce travail est réalisé dans le cadre d’une collaboration avec 3 laboratoires :
- Le Laboratoire de “Biomaterials Laboratory, Martinos Center, Massachusetts
General Hospital and Harvard Medical School Boston, MA, USA”.
- Le Laboratoire de Chimie de la Matière Condensée - Collège de France, Université
Pierre et Marie Curie, Paris VI, France.
- Le Laboratoire de Physique de la Matière Condensée (PMC), Ecole Polytechnique,
France.
En tant que lauréat Fulbright, je tiens à exprimer mes vifs remerciements et toute ma
reconnaissance à tous les membres de la Commission Maroco-Américaine (MACECE) « The
Moroccan-American Commission for Educational and Cultural Exchange » et AMIDEAST
avec le Fulbright Scholarship Program, pour le support financier, le suivi et leur soutien le
long de mes séjours aux USA dans le cadre de la préparation de cette thèse.
Je suis particulièrement reconnaissante à Monsieur Jerome L. ACKERMAN Director
of Biomaterials Laboratory, Martinos Center, Massachusetts General Hospital and Professor
of Radiology at Harvard Medical School in USA, de m’avoir gentiment accueilli parmi son
équipe. Je le remercie d’avoir cru à mon projet et d’avoir mis à ma disposition tous les
moyens nécessaires pour le mener.
Milles merci à Mr Thibaud Coradin, Directeur de recherche au CNRS, et à M
me
Sylvie
Masse chargée de recherche scientifique au Laboratoire de Chimie de la Matière Condensée
de Paris Université Pierre et marie Curie Paris VI- France, pour leur collaboration et l’accueil
chaleureux qu’ils m’ont réservé durant mes séjours à Paris. Qu’ils trouvent ici l’expression de
ma respectueuse gratitude pour leurs précieux conseils et leurs discussions fructueuses.
Un grand merci au Dr. Khalid LAHLIL de l’Ecole Polytechniques de Paris – France
pour sa collaboration scientifique, pour l'aide apportée durant mes expériences et pour
l’intérêt constant qu’il a porté à ce travail.
Mes remerciements les plus sincères s’adressent au président du jury Monsieur le
Professeur Abdallah ZRINEH de la Faculté des Sciences, Rabat – Maroc de m’avoir fait
l’honneur de présider le jury de cette thèse. Ses discutions scientifiques intéressantes dans le
suivi et la réalisation de mes travaux de recherche ont été d’une qualité supérieure.

Je suis sensible à l’honneur que me fait Monsieur Jamal NAJA, Professeur et Vice
Doyen à la Faculté des Sciences et Techniques de Settat (FSTS), en acceptant d’être
rapporteur de ce travail et de faire partie du jury. Qu’il trouve ici l’expression de ma
respectueuse gratitude.
Mes remerciements s’adressent à Monsieur Mohamed HAMAD, Professeur à la
Faculté des Sciences, Rabat – Maroc autant que rapporteur de cette thèse et pour sa présence
parmi les membres du jury, afin de juger le contenu de ce travail.
Je suis honoré que Monsieur Thibaud. CORADIN, Directeur de recherche au CNRS et
responsable de l’équipe de recherche « Biomatériaux et environnement » au Laboratoire de
Chimie de la Matière Condensée de Paris – France, ait accepté d’être rapporteur et membre du
jury de ce travail. Qu’il trouve ici l’expression de ma respectueuse gratitude.
Mes remerciements s’adressent à Monsieur Jerome L. ACKERMAN, Director of
Biomaterials Laboratory, Martinos Center, Massachusetts General Hospital and Professor of
Radiology at Harvard Medical School in USA d’avoir aimablement accepté de se déplacer
pour juger ce travail malgré tous ses engagements et ses nombreuses occupations.
Je tiens à remercier Monsieur Philippe BARBOUX, Professeur et responsable des
relations industrielles à l’Ecole Nationale Supérieure de Chimie de Paris- France, pour le
grand honneur qu’il m’a fait en acceptant d’être membre du jury de cette thèse.
Mes remerciements s’adressent à Monsieur Abdelaziz LAGHZIZIL pour l’attention
constante avec laquelle il a suivi mes travaux de recherche et pour ses précieuses discussions
scientifiques, qu’il soit également très vivement remercié d’avoir accepté d’être membre du
jury.
De l’autre bout de la terre, mes meilleurs sentiments d’amitié s’adressent :
- A tous les membres du Laboratoire de Chimie de la Matière Condensée (LCMC),
Paris VI, Jussieu France, et en particulier, G. Laurent, I. Genois, N. Abdoul-Aribi, M.
Selmane et L. G. Patrick, qui ont été toujours là pour m'aider au cours de mes stages à
l’Université Pierre et Marie Curie, Paris VI France.
- A tout le personnel de Martinos Center et Harvard Medical School Boston - USA, et
en particulier, Messieurs Wu, Yaotang, Choukri Mekkaoui, David Alcanta, Dionyssios
Mintzopoulos et M
me
Haihui Cao.
- Aux responsables du « Fulbright Student Exchange Program » à Washington DC -
USA, notamment, M
me
Stephanie Whitlatch et M
me
Elisa O’Keefe pour leur politique de
proximité et leur compétence.
Je tiens à remercier les membres de l’UATRS-CNRST, en particulier Messieurs B. Jaber, K.
Anouar, M. Benissa et M
lle
H. Oauddari, pour leur aide technique à la caractérisation et à
l’analyse chimiques de quelques produits présentés dans ce travail.
Je souhaite associer à ces remerciements tous ceux qui ont contribué à la réalisation de
ce travail en particulier à tous mes compatriotes et mes amis qui ont rendu mes séjours en
Amérique et en France si agréables, plein d’encouragement, d’espoir et de compréhensions,
aux enseignants-chercheurs et tous mes ami(e)s et collègues du Laboratoire de Chimie
Physique Générale et du Département de Chimie de la Faculté des Sciences de Rabat - Maroc,

de Martinos Center, MGH et Harvard Medical School de Boston - USA et de LCMC et
UPMC de Paris - France.
Je désire terminer en remerciant mes sources de joie, de bonheur et de force à mes
parents, ma sœur Sarah, mon frère Yahya et mes deux sœurs jumelles Sawsane et Fadwa qui
ont joué évidemment un très grand rôle dans cette thèse. Leur soutien moral et matériel m’a
été indispensable tout au long de ces années. J’espère que ce travail leur témoigne toute ma
reconnaissance et mon estime à leurs égards. Merci de m’avoir soutenue et encouragé pendant
les moments de doutes.

Sommaire
Introduction générale
……………………………………………………………..
1
Partie I : Généralités sur la gestion des métaux lourds et leur élimination
par des solides poreux et fonctionnalisés ……………………
5
Chapitre I : Comportement physico-chimique des métaux dans l’environnement ….
7
Introduction …………………………………………………………………………..
8
I. Toxicité des métaux et leurs conséquences sur les écosystèmes…………………… 8
II. Sources et devenir des contaminants métalliques dans l’environnement…………. 9
II.1. Milieu aquatique………………………………………………………………… 9
II.2. Contamination des sols par les métaux lourds et remédiation…………………. 10
III. Physico-chimiques des réactions à l’interface solide – liquide………………….. 11
III.1. Hydrolyse du métal : cas des ions Pb
2+
et Zn
2+
……………………………….. 11
III.2. Complexation des ions métalliques aqueux…………………………............... 13
III.3. Interactions avec les carbonates et les phosphates……………………………. 14.
III.4. Interactions avec les amines et carboxylates………………………………….. 14
III.5. Immobilisation des cations métalliques par une surface……………………… 15
III. 6. Mécanismes d’interaction solide – métal……………………………………. 16
III.6.1. Adsorption …………………………………………………………………… 16
III.6.1. 1.Adsorption non spécifique………………………………………………… 16
III.6.1.2. Adsorption spécifique………………………………………………………. 17
III.6.2. Echange ionique………………………………………………………………. 18
III.6.3. Précipitation à la surface………………………………………………….. 18
IV. Elimination des métaux lourds par les solides poreux ………………………….. 19
IV.1. Les argiles…………………………………………………………………….. 19
IV.2. Les charbons actifs…………………………………………………………… 19
IV.3. La silice poreuse et ses dérivées………………………………………………. 20
IV.4. Les phosphates de calcium……………………………………………………. 20
Conclusion………………………………………………………………………….. 21
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
1
/
166
100%