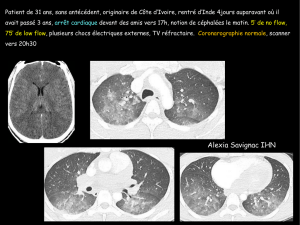
Cardiomyopathie ventriculaire droite arythmogène

Définitions
Mort subite
Mort naturelle, inattendue, survenue dans
l’heure qui suit l’apparition de symptômes
Guidelines for Autopsy Investigation of Sudden Cardiac Death.
C. Basso, M. Burke, P. Fornes, P. Gallagher, M. Sheppard, G. Thiene,
A. van der Wal, on behalf of the
Association for European Cardiovascular Pathology
Virchows Archiv, 2007

Définitions
Trépied électrophysiologique
de la mort subite
Substrat arythmogène
“Trigger”
Ischémie
Hypoxie
Troubles métaboliques
Troubles hydroélectrolytiques
Toxique
Système sympathique

Pathologie coronarienne

Mort subite coronaire
−Au moins une des artères coronaires
épicardiques présente une sténose > 75 %
.Plaque athéroscléreuse,
.Thrombose / hématome intra-plaque,
.Dissection,
.Vascularite.
−Occlusions coronaires intramyocardiques
−Diagnostic d’exclusion

Thromboses coronaires
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
1
/
51
100%