phénylcétonurie n - Raymond Rodriguez SVTperso

La phénylcétonurie (ou PCU)
La phénylcétonurie est un trouble héréditaire qui se caractérise par une arriération mentale accompagnée d’un re-
tard du développement physique, de troubles neurologiques et du comportement et d’un déficit pigmentaire. En France elle
touche un individu sur 16 000.
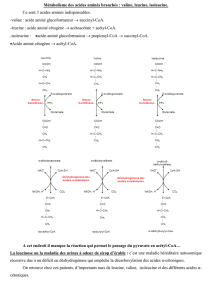
Elle est due à l’absence d’une enzyme (la phénylalanine hydroxylase ou PAH) qui permet de transformer la phé-
nylalanine (PA, qui est un acide aminé indispensable présent dans la plupart des protéines animales et en particulier le
lait) en tyrosine (un autre acide aminé). Cette dernière peut ensuite être transformée en thyroxine (hormone de la thy-
roïde), adrénaline (une hormone) et mélanine (qui pigmente la peau). Le déficit en PA hydroxylase entraîne donc une accu-
mulation de la PA et une diminution des métabolites normaux ce qui provoque les symptômes de la maladie. Cependant,
l'apport de tyrosine alimentaire est suffisant pour que la tyrosine ne soit pas diminuée dans la PCU.
Le dépistage se fait dès la naissance (vers le 6ème jour) en mesurant le taux de PA dans le sang. Si le taux de PA est
élevé, et si la PCU est confirmée, l'enfant est pris en charge le plus rapidement possible.
Le traitement est uniquement diététique et nécessite un apport restreint de phénylalanine dans un régime stricte-
ment établi. Cette alimentation spécialisée coûte cher mais elle est entièrement gratuite pour les familles qui la reçoivent
directement de la Pharmacie Centrale des Hôpitaux de Paris.
L'acceptation de ces aliments diététiques est facile au cours des six premiers mois. Par la suite, ils peuvent être as-
sociées à des aliments naturels et à des produits diététiques hypoprotidiques qui évitent la monotonie, cause d'anorexie
et de chapardage. Mais l'apport d'une quantité suffisante de protides accompagnés d'une quantité limitée de PA restera
toujours un problème car l'acceptabilité de ces produits diététiques, surtout chez les plus grands, dont le goût est diversifié,
n'est pas toujours bonne, et ceci malgré des tentatives de présentations plus agréables pour l'enfant : liquide ressemblant à
du jus d'orange ou solide ressemblant à des barres de chocolat. C'est pourquoi l'aide d'une diététicienne, en contact direct
avec la famille, est de la plus grande importance.
La durée du traitement varie selon les auteurs de 6 à 12 ans. Les enfants atteints de PCU doivent être régulièrement
suivis et leur développement mental et intellectuel surveillé par des examens psychologiques pratiqués à intervalles régu-
liers. Les résultats des traitements sont bons. Un traitement bien suivi évite l'arriération mentale et, débuté précocement,
permet un développement mental et intellectuel normal. Toutefois les résultats scolaires, qu'on commence à connaître, sont
moins bons et il existe fréquemment des troubles d'apprentissage du langage, une imprécision motrice, des tremblements.
Enfin, les progrès réalisés dans le dépistage et le traitement précoce font que les filles atteintes de PCU, ayant une intelli-
gence normale, atteignent la puberté, vont se marier et, ayant une fécondité normale, vont avoir des enfants.
Mais deux enquêtes ont montré qu'au cours de ces grossesses, il y avait beaucoup d'avortements (20 à 25 %), un re-
tard mental (72 à 75 %) et des malformations diverses (microcéphalie 65 %, retard de croissance 36 %, malformations car-
diaques 12 %). Le nombre de femmes dans ce cas augmentant, on risque de se retrouver devant un nombre de nouveau-
nés de mères phénylcétonuriques égal au nombre de nouveau-nés dépistés. C'est dire l'importance des recherches entre-
prises pour prévenir cette nouvelle pathologie.
Ceci pose donc le problème d'un traitement des femmes phénylcétonuriques désirant avoir un enfant : un régime
pauvre en PA débutant dès avant la conception est actuellement conseillé, il est indispensable pendant la grossesse pour
que l'enfant ne soit pas microcéphale. On doit naturellement maintenir des apports caloriques normaux et des apports proti-
diques ; là encore se posent des problèmes d'acceptabilité du régime.
Le diagnostic anténatal est maintenant possible grâce aux progrès des méthodes de biologie moléculaire, car le
gène a été cloné. Mais il pose un problème d'éthique : le diagnostic prénatal, devant conduire à proposer éventuelle-
ment l'interruption de grossesse, est-il légitime pour une maladie traitable ?
D’après http://www.med.univ-rennes1.fr/etud/pediatrie/erreurs-metabolisme.htm
1
/
1
100%