10 Les dérivés carbonylés

Les dérivés carbonylés
A - Propriétés chimiques
Ce sont des dérivés qui possèdent un groupement C = O
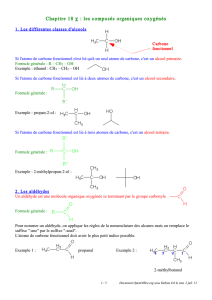
On distingue les aldéhydes, un des substituant de l’atome de carbone étant un
hydrogène, le deuxième pourra être un H (méthanal) ou un alkyle, le groupement
aldéhydique est toujours site en bout de chaine,
Dans le cas d’un dérivé cétonique, ce sont 2 groupements alkyles qui sont fixé sur
l’atome de carbone, donc le groupement Carbonylé d’une cétone sera toujours situé à
l’intérieur de la chaine.
Quelques caractéristiques :
Il s’agit d’une molécule plane puisque le carbone de même
que l’oxygène est hybridé sp2 avec des angles de liaisons
très voisin de 120°, cette double liaison carbone – oxygène
peut être comparé à la double liaison C = C d’un composé éthylénique, toute fois il existe
un certains nombre de différence :
1er il s’agit d’une liaison plus courte que C = C, donc on aura entre le carbone et
l’oxygène une densité électronique supérieur
2e cette double liaison C = O est plus forte qu’une liaison C = C
3e cette liaison est fortement polaire ceci pouvant être montré
expérimentalement par la mesure d’un moment dipolaire élevé.
Puisque le carbone et l’oxygène sont hybridé sp2, ils possèdent tout les deux une
orbitale non hybridé p possédant un électron et c’est le recouvrement latéral de ces
orbitale p (perpendiculaire au plan de la molécule) qui va constitué la liaison pi
L’oxygène possède 2 orbitales hybridés sp2 possédant chacune 2 électrons qui sont
situé dans le plan de la molécule et qui occupent des directions qui sont celles des
substituant dans le cas des dérivés éthyléniques. Les paires électroniques sont bien
dégagées, très accessible aux réactifs, donc C = O peut intervenir par les doublets
d’électrons libres de l’atome d’Oxygène
La différence d’électronégativité entre le carbone et l’oxygène, va entrainer une
déformation du nuage d’électrons pi avec une densité électronique plus forte du côté de
l’atome le plus électronégatif (O), il portera une charge O-, le carbone portant une charge
positive (déficit électronique).

Au point de vu réactivité ceci se traduit par une attaque
nucléophile du carbone de la double liaison C = O et une
attaque électrophile de l’atome d’oxygène, on verra qu’il
s’agit presque essentiellement de protonation de l’atome
d’oxygène, ce qui entraine la formation d’un carbocation.
Si on considère les aldéhydes et les cétones.
Les aldéhydes, il n’y a qu’un groupement alkyle et 1 H, donc
l’approche du réactif nucléophile va être plus facile dans le
cas des aldéhydes que dans le cas des cétones puisque
l’encombrement stérique de la double liaison C = O est moins important pour les
aldéhydes que pour les cétones.
2e type de propriété due à la mobilité de l’hydrogène porté par le carbone en α du
groupement carbonyle, en effet par suite de l’existence d’une charge positive sur ce
carbone, celui ci va attiré les électrons des liaisons voisines et notamment ceux de la
liaison carbone – Hydrogène, diminuant la densité entre le carbone et l’hydrogène donc
favorisant son départ sous forme de proton avec formation d’un carbanion.
Dans le cas des aldéhydes et cétones on a un pKa de l’ordre de 19 – 21 ce qui signifie que
ce composé est moins acide que les alcools.
La 2e raison de la mobilité de l’hydrogène est que le carbanion
formé va être stabilisé par mésomérie avec transfert de la charge
négative sur l’atome d’oxygène qui s’accommode mieux de cette
charge négative.
D’un côté on a un carbanion, de l’autre on a un énolate qui est un sel d’enol
Rappel : la tautomérie céto énolique
Dans ce cas là il s’agit d’un équilibre puisqu’il existe des cas où les 2 composés peuvent
être isolés et existent réellement. Qui dit tautomérie dit déplacement d’électrons et
déplacements d’atomes (mésomérie = déplacement d’électrons).

Une particule électrophile va pouvoir s’additionner sur le carbanion, c’est la majorité des
cas, mais il existe des cas particuliers ou la particule électrophile s’additionne sur
l’oxygène.
A partir des dérivés carbonylés on pourra avoir des réactions de réduction pour aller
aux alcools et des oxydations pour arriver aux acides carboxyliques.
B – Réaction d’additions nucléophile
1°) réaction qui conduisent à des produits stables (qui ne seront pas le siège
d’éliminations ultérieur)
a) l’eau
OH – s’addition sur C + et H + sur O – on obtient 2 composé de 2 fonction hydroxyle
porté par le même carbone, c’est donc un Diol, et
comme les 2 hydroxyles sont sur le même
carbone, on les appel gem diol(jumeaux)
Dans le cas des cétones ce n’est pas très équilibré
car OH est faiblement nucléophile et que C est
faiblement polarisé donc on va augmenter la
polarisation en mettant sur le carbone en α des
substituant fortement électro attracteur, la
présence de ces substituant augmente la polarité
C+ de C = O et quoi que le nucléophile soit faible,
il va s’additionner sur le carbone fortement
polarisé, d’où avec ses groupements électro
attracteurs, la flèche va vers la formation du gem diol.
b) action des alcools : réaction d’acétalisation
On obtient un acétal ou hémi acétal
Il nous faut un catalyseur car le C est pas asse polarisé, si on
met des protons dans le milieu, il va s’additionner sur O et le
carbone devient un carbocation, le carbone est donc
suffisamment polarisé pour qu’une molécule d’alcool par
l’intermédiaire d’un des doublets libre du groupement
hydroxyle vienne s’additionner sur le carbone.
Cette réaction d’acétalisation nécessite une catalyse acide qui
par addition sur le groupement carbonylé entraine la formation
d’un carbocation. La molécule d’alcool par l’intermédiaire d’un
des doublets libres de l’oxygène se fixe sur ce carbocation

entrainant la formation intermédiaire d’un oxonium qui perd spontanément un proton
avec formation d’un hémiacétal.
L’hémi acétal possède un groupement OH, donc élimination d’eau, qui forme un
carbocation et fixation d’une nouvelle molécule d’alcool
Réaction importante car permet d’expliquer la structure cyclique des oses, il y a
formation d’un hémi acétal par réaction du groupement carbonylé d’un aldose ou d’un
cétose sur le groupement OH porté par le même ose
c) action des organomagnésiens
Les organomagnésiens ont pour formules RMgX et étant donné l’électropositivité de
l’atome de magnésium il sera toujours polarisé δ + et R δ -, donc lorsqu’on met un
organomagnésien en présence d’une double liaison polarisée comme c’est le cas pour la
liaison carbonyle, on observe une double addition de
MgX δ + sur O δ – et de R – sur le C du groupement
carbonyle, on obtient donc un composé d’addition
qui correspond en fait à un alcoolate de magnésium,
ces composés d’additions sont des composés très
sensibles à l’hydrolyse et on va avoir l’hydrolyse de la liaison O – Mg avec formation d’un
groupement hydroxyle (fonction alcool) et de MgX- OH, on laisse tombé MgX – OH. Il
s’agit donc d’un moyen de transformer une fonction cétonique en un alcool.
Le plus simple des dérivés carbonylés est le formaldéhyde, les 2 liaisons restantes sont
occupées par un atome d’hydrogène et quand on fait agir un magnésien sur le
formaldéhyde on obtient un alcool Primaire et c’est la seule façon d’obtenir un alcool I.
2e cet alcool possède un atome de carbone de plus que le magnésien de départ, il faut
compté le nombre de carbone du composé de départ et le nombre de carbone du
composé d’arrivé
2e) réaction d’un organomagnésien avec un autre aldéhyde,
on obtient un alcool II
3e) l’action d’un magnésien sur une cétone donne un alcool III

2e) réactions d’additions nucléophiles suivies de réactions d’élimination
(intermédiaires instables)
a) NH3 et amines
L’ammoniaque ou ses dérivés vont s’additionné par
l’intermédiaire du doublet libre de l’atome d’azote
par le C δ + du dérivé carbonylé, on obtient un
composé intermédiaire instable qui perd
spontanément une molécule d’eau pour donner un
composé possédant une liaison C=NH qui est une
imine on parlera d’aldimine lorsque le composé
carbonylé est un aldéhyde et de cétimine lorsque ce sera une cétone.
b) Dérivés de l’ammoniac : Z – NH2
On a substitué un Hydrogène de l’ammoniac par un groupement Z.
Pour que la réaction marche bien, il faut transformer le carbocation C δ+ et en C+,
l’addition de l’atome d’azote par son doublet
libre se fera plus facilement car la charge sur
l’atome d’azote sera plus importante, on aura
donc une catalyse acide (H+), H+ va se fixer
sur O- et on obtient un composé d’addition
qui par mésomérie va donner le carbocation
Ce carbone est fortement chargé, l’addition
nucléophile se fait d’autant plus facilement
pour donner comme précédemment ce composé d’addition
La 2e étape consiste en l’élimination d’une molécule
d’eau, le mécanisme est simple, on est en milieu acide
donc (H+) dans le milieu, le composé intermédiaire à
une fonction OH qui attire un proton pour former un
ion oxonium, l’ion oxonium est instable et perd
facilement une molécule d’eau pour donner un
carbocation qui perd à son tour un hydrogène sous
forme de protons et donne des dérivés possédant une
double liaison carbone – azote (C = N).
Il faut une petite quantité de Protons (quantité catalytique) pour la formation du
premier carbocation mais il ne faut pas qu’il y ai trop de protons sinon il se fixeraient sur
le doublet libre de l’atome d’azote empêchant l’addition nucléophile sur le carbocation.
 6
7
8
6
7
8
1
/
8
100%