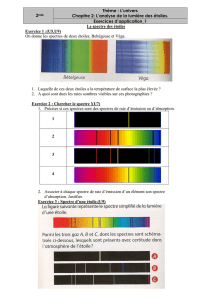
spectroscopie uv

1
SPC5 UEF1
COURS/TD SPECTROSCOPIES
SPECTROSCOPIES MOLECULAIRES
(UV - visible, IR, RMN de 1H et 13C, masse) :
Application à l’élucidation structurale des composés organiques.
(cours de M. LE BLANC)
La spectroscopie correspond à l’étude du rayonnement électromagnétique
émis, absorbé ou diffusé par des atomes ou des molécules. L’interaction de la
matière avec le rayonnement électromagnétique se fait généralement par
l’intermédiaire de sa composante électrique.
SPECTROSCOPIE ATOMIQUE :
La spectroscopie atomique a joué un rôle important dans le développement de
la théorie quantique, mais le nombre des atomes est limité et les ions possédant plus
de trois ou quatre charges sont difficiles à obtenir. La plupart des spectres sont déjà
connus et interprétés. Ils gardent cependant un intérêt en astronomie, en chimie
analytique et en physico - chimie du solide, notamment pour l’étude de nouveaux
matériaux inorganiques.
LE SPECTRE DE L’HYDROGENE :
Il nous permettra d’énoncer quelques définitions et principes fondamentaux :on
sait que la théorie quantique, qui s’applique aux atomes et aux molécules, nous dit
qu’ils ne peuvent exister que dans des états dont l’énergie a une valeur discrète E,
liée à certains paramètres quantiques. L’exemple le plus connu est le spectre
d’émission de l’atome d’hydrogène, dont l’observation a permis d’illustrer la théorie
quantique : c’est un spectre de raies, dont le nombre d’onde est déterminé par :
n1 < n2 correspondent à deux valeurs du nombre quantique principal n de l’atome
d’hydrogène et RH à la constante de Rydberg, ce qui traduit la condition de fréquence
de Bohr : E2 - E1 = h, qui traduit que l’énergie totale du système doit être
conservée quand se produit une transition.
La précision E avec laquelle on peut connaître l’énergie E d’une molécule
ou d’un atome est reliée au temps d’observation t par le principe d’incertitude :
E.t > h/2. On voit que la précision sera encore très bonne tant que t sera
supérieure à 10-10 s.
Le principe d’incertitude aura des répercussions sur la largeur des bandes
spectrales.
L’état fondamental d’une molécule ou d’un atome est l’état le plus peuplé
lorsque, le matériau étant en équilibre thermique, on le refroidit progressivement
jusqu’au zéro absolu, sans qu’il y ait modification chimique. Tous les états d’énergie
supérieure sont dits états excités.
=
_
1
_
=RH(n1
2
_
1-_
1
n2
2)

2
Soit A l’état fondamental et B, C,… les états excités. Pour décrire une
transition, on place toujours l’état d’énergie le plus élevé à gauche. Ainsi plusieurs
processus peuvent se produire lors de l’interaction de :
B A correspond à l’absorption d’un rayonnement correspondant à la
transition de l’état fondamental vers un état excité.
B A correspond à l’émission d’un rayonnement correspondant à la
transition d’un état excité vers l’état fondamental
B A indique que la transition a lieu dans les deux sens.
SPECTROSCOPIES MOLECULAIRES : INTERACTIONS RADIATION - MOLECULE :
Le nombre de molécules est très grand et ne cesse de grandir. Les
spectroscopies moléculaires continuent donc à se développer.
Des radiations de longueur d'onde très variable (cf. Fig. 1) peuvent induire des
changements dans les mouvements ou la structure des molécules. Ces interactions,
détectées et mesurées dans un spectromètre, sont matérialisées sous forme de spectres
(UV, visible, IR, RMN, masse...).
Longueurs d'onde ( ) croissantes
Rayons
X UV
lointain Proche
UV Visible Infrarouge Microondes Ondes
radio RMN
200 nm 400 nm 800 nm
2,5 m20 m5 m
1 m
Transitions
électroniques Vibrations
moléculaires Rotations
moléculaires Spins
nucléaires
Fréquences (énergies) décroissantes
Fig. 1: Zônes du spectre électromagnétique utilisés dans l'élucidation des structures des
composés chimiques et nature des phénomènes mis en jeu.
La plupart des états énergétiques d’une molécule (rotation, vibration, spin
nucléaire, répartition électronique sur les orbitales moléculaires) étant quantifiés, un
changement d’état (transition) se traduira par l’absorption, ou l’émission d’une radiation
quasi - monochromatique.

3
PRINCIPE DE LA SPECTROSCOPIE :
La spectroscopie est une technique permettant d’enregistrer l’intensité d’une
radiation selon sa longueur d’onde (ou sa fréquence ) : une brusque variation de
cette intensité (qui correspond à une absorption ou à une émission de radiation)
indiquera l’existence d’une telle transition. Ceci se traduira sur l’enregistrement, du
type : I (intensité) (ou A absorbance, ou T transmittance) = f () (ou
appelé spectre, par l’apparition selon les cas:
- d’une bande fine pour une certaine valeur de ou (RMN, IR, microondes)
- d’une bande large centrée autour d’une certaine valeur de ou (UV-visible)
La valeur de cette longueur d’onde (ou de cette fréquence) est une
caractéristique intrinsèque de la substance. Sa mesure sera un indice qui contribuera
à son identification.
Dans la plupart des spectroscopies, on travaille en absorption :
- on fait traverser la substance à étudier par un rayonnement
- on compare, pour une même valeur de (ou ) l’intensité du rayonnement
émergent I à l’intensité du rayonnement incident I0. Quand I/I0 < 1 on a
absorption.
Les dispositifs expérimentaux permettant de réaliser cette comparaison seront
éventuellement décrits ultérieurement.
HISTORIQUE DE L’ELUCIDATION STRUCTURALE EN CHIMIE ORGANIQUE:
Dès le commencement de la chimie organique s’est posé le problème de la
détermination de la structure des composés.
Avant les années 40, la méthodologie suivante était utilisée :
- On cherchait d’abord à savoir si le composé était déjà connu en comparant
certaines de ses propriétés physiques (points d’ébullition, de fusion, indice de
réfraction, densité, etc…) avec celles déjà tabulées dans la littérature.
- Si le composé était inconnu :
- il fallait d’abord le purifier le (par distillation, recristallisation,…)
- s’assurer de sa pureté par le peu de méthodes dont on disposait (points de
fusion et d’ébullition nets par exemple)
- on cherchait ensuite à déterminer sa formule brute, à partir de mesures de
masse molaire et d’analyse centésimale
- on cherchait ensuite à mettre en évidence la présence de groupes
fonctionnels par des tests caractéristiques (eau de brome, liqueur de
Fehling, Lucas, etc..) ou l’obtention de dérivés caractéristiques (2,4-DNPH,
sels de S-benzylthiouronium,…)
Depuis, sont apparues de nombreuses méthodes de séparation et de purification
beaucoup plus efficaces (chromatographies), la masse moléculaire, voire la formule
brute peuvent être déterminées par spectrométrie de masse, la spectrométrie de
résonance magnétique nucléaire (RMN) de 13C permet de préciser la structure du
squelette carboné, la présence de groupes fonctionnels peut être mise en par
spectroscopie infrarouge (IR) ou de RMN (1H, 13C). Le principe et quelques
applications de ces différentes spectroscopies seront brièvement décrites ci-après.

4
CARACTERISTIQUES DES MOLECULES ORGANIQUES:
- Elles contiennent tous du carbone, et possèdent généralement des liaisons
entre atomes de carbone
- La plupart contiennent des atomes d'hydrogène souvent, mais pas toujours
liés à ces atomes de carbone
- Elles contiennent souvent d'autres éléments, appelés hétéroatomes, présents
en nombre limité: O, N, S, P, halogènes, généralement liés au carbone, quelquefois liés
entre eux.
- La plupart contiennent un groupe fonctionnel, défini comme un groupe
d'atomes liés entre eux autrement que par des liaisons simples C-C ou C-H.
COMPLEMENTARITE ET POINTS FORTS DES DIFFERENTES METHODES SPECTRO -
SCOPIQUES :
INFRAROUGE:
Permet surtout de voir quels groupes fonctionnels sont présents.
Peut donner quelques indications sur le squelette carboné (ramifi-
cations par ex)
RMN DU PROTON:
Permet de définir l'environnement proche de chaque groupe
d'atomes d'hydrogène de la molécule: on peut ainsi dire à quel type d'atome de
carbone chaque atome d'hydrogène est attaché, et quel hétéroatome ou groupe
fonctionnel est à proximité.
RMN DU CARBONE 13:
Permet de décrire le squelette carboné de la molécule.
Permet de voir si des atomes de carbone ne sont pas liés à des atomes
d’hydrogène
SPECTROMETRIE DE MASSE:
Peut permettre de déterminer la masse moléculaire du composé et de
certains de ses fragments.
En haute résolution, permet même de proposer la formule brute du
composé et de ses fragments
Il est rarement possible de déterminer la structure d'un composé organique à
partir d'un seul type de spectroscopie. En pratique, plusieurs spectres sont souvent
nécessaires, d'où l'utilité d'envisager chaque type de spectroscopie en relation avec
les autres, ce qui évite de trop vouloir tirer d'informations d'un seul spectre.
L’identification de la structure d’un composé organique s’apparente à une
enquête policière où on accumule des indices convergents à l’aide d’une logique
« floue », c’est à dire n’utilisant pas (ou rarement) de relations mathématiques.
La stratégie la plus efficace - c’est à dire l’ordre dans lequel les différentes
spectroscopies doivent être utilisées - vous est donné ci-dessous. On peut quelquefois
sauter des étapes dans le cas de molécules simples. Avant l’emploi de toute méthode
spectroscopique, il faut s’être assuré de la PURETE DE L’ECHANTILLON, par la
mesure de certaines constantes caractéristiques (points de fusion, d’ébullition) et

5
l’application de méthodes de purification adaptées: distillation, recristallisation,
chromatographies,...
On enregistre et on interprète alors les spectres obtenus dans l’ordre suivant:
1) SPECTRE DE MASSE
Formule brute du composé si haute résolution
2) SPECTRE INFRAROUGE
Identification de groupes fonctionnels
3) SPECTRE DE RMN DE 1H
Confirmation du nombre et du type d’atomes d’hydrogène présents
Identification de la connectivité des goupes hydrocarbonés
4) SPECTRE DE RMN DE 13C
Confirmation du type d’atomes de carbone présents
Confirmation du nombre et du type de groupements possédant des
liaisons C-H
PROPOSITION D’UNE STRUCTURE
5) RETOUR SUR LE SPECTRE DE MASSE
Recherche et identification de fragments présents dans la structure
proposée
CONFIRMATION/INFIRMATION DE LA STRUCTURE
 6
7
8
9
10
11
12
13
14
15
6
7
8
9
10
11
12
13
14
15
1
/
15
100%