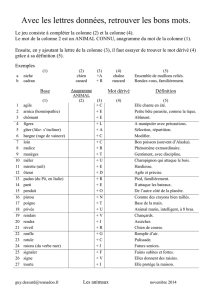
Présentation de l`atelier de génie chimique de l`ENCPB

Udppc PARIS de Sciences Atelier de génie chimique
ENCPB Oct 07 S. Bru, C. Canu, C. Kressmann
1
PRESENTATION DE
L’ATELIER DE GENIE CHIMIQUE DE L’ENCPB
I. Généralités
L’atelier de Génie Chimique de l’ENCPB est unique en France de par sa conception qui utilise du matériel
industriel en demi-grand. Ce type de matériel permet d’appréhender tous les problèmes techniques qui
peuvent se poser lors d’une opération unitaire, avant de concevoir l’installation en grand. Il suit la
validation du procédé défini au laboratoire.
II. Historique
En 1975, le Directeur de l’ENCPB demande à la chambre syndicale des industries chimiques une
subvention pour monter un atelier de génie chimique. Une somme de 1 000 000 F doit être versée sur 3
ans. Des enseignants sont alors chargés de dessiner les plans des installations. Ils s’inspirent du
matériel déjà utilisé au niveau BEP.
Au bout de 3 ans, la chambre syndicale souhaite voir les installations. Rien n’ayant été construit, Rhône
Poulenc (aujourd’hui devenu Aventis), gros participant dans cette opération, décide d’envoyer à mi-
temps un de ses ingénieurs expérimentés, Monsieur Paul ANGLARET, pour démarrer le projet en
collaboration avec l’usine de Vitry. Il est alors nommé chef des travaux.
Le projet démarre réellement en 1975. S’appuyant sur des schémas de principe pré-établis, le bureau
d’études de Rhône Poulenc fournit les plans de chaudronnerie, de tuyauteries, d’implantations, etc.
Un appel d’offres est lancé pour la première tranche de travaux. Elle comprend deux unités de
fermentation, deux installations d’extraction (liquide-liquide et liquide-gaz), deux unités de
rectification (continue et discontinue), une unité de cristallisation filtration, une unité en émail pour la
fabrication de produits chimiques en milieu corrosif, deux unités couplées de fabrication polyvalente,
une unité de transfert de chaleur et une de centrifugation.
Les travaux sont lancés en janvier 1976 et doivent être terminés rapidement. Mr ANGLARET
embauche alors un collaborateur, Mr Kazmierzak, pour l’aider dans la réalisation de son projet. Arrivé
en février 1976, ce dernier suit les travaux, fait les essais à l’eau et dessine les installations de la
deuxième tranche de travaux. Les installations accueillent pour la 1° fois des élèves de BTS Chimistes
2° année en septembre 1976.
Mr Anglaret obtient ensuite une nouvelle subvention pour concevoir des installations simples et à
caractère répétitifs plus faciles à utiliser par des élèves débutants : installations d’étude des
équilibres liquide-liquide et des équilibres liquide vapeur, de la fluidisation, des pertes de charge en
colonne et une unité d’hydrodynamique de l’air. En 1977–1978 la deuxième tranche de travaux est
construite avec les essais des manipulations, les modifications à apporter aux installations, au protocole
des travaux pratiques etc.
C’est à cette époque et avec la participation de Mr Anglaret, représentant de la chambre syndicale, de
l’inspection générale et de l’ancien directeur de l’ENCPB, Mr. Yves TALLET, que le génie chimique
apparaît dans les textes de programme à la place de la technologie.
Ensuite, de nouveaux plans d’installations de fabrication polyvalente ont été définis, dessinés et prévus
pour suivre une progression pédagogique et accueillir des élèves de terminales STL (anciennement F6).
Quand l’automatisation et le développement de l’informatique ont commencé dans les années 85-86,
l’atelier s’est vu équipé d’une installation supplémentaire, une colonne de distillation automatisée.
La dernière acquisition est une unité d’évaporation double effet.

Udppc PARIS de Sciences Atelier de génie chimique
ENCPB Oct 07 S. Bru, C. Canu, C. Kressmann
2
TP1 : Estérification dans un réacteur émaillé
I. La réaction d’estérification
Le poste « chimie email » permet de préparer un ester (l’acétate de butyle) par réaction de l'acide
acétique sur un alcool primaire (butanol) en présence d’une catalyse acide (acide sulfurique) :
H+
CH3COOH + C4H9OH = CH3COOC4H9 + H2O
On utilise un réacteur en email car les acides utilisés présentent un caractère hautement corrosif.
II. Accélération de la réaction (étude cinétique)
Cette réaction est assez lente (pour atteindre le rendement maximal, il faudrait plusieurs mois). On
cherche donc des moyens d'accélérer la réaction :
- Augmentation de la température : si elle n'a aucune influence sur le rendement, elle améliore
grandement la cinétique. On utilise un chauffage à la vapeur d’eau (sous P= environ 4bar) dans une
double enveloppe.
- Utilisation d'un catalyseur : on introduit pour cela un acide fort (H2SO4), qui permet d'augmenter le
caractère électrophile du groupe carboxyle porté par l’acide carboxylique.
III. Optimisation du rendement (étude thermodynamique)
Le rendement d’une estérification dépend très peu de la nature de l'acide carboxylique utilisé. Il
dépend surtout de la classe de l'alcool utilisé : pour des réactifs introduits en quantités équimolaires, il
est de 67 % avec un alcool primaire (butanol dans notre cas).
Voici donc nos possibilités :
- Mettre le butanol en excès (réactif le moins coûteux). L’équilibre est alors déplacé vers le sens
direct d’après la loi de Le Chatelier.
- Eliminer l’ester ou l’eau au fur et à mesure de leur formation. On choisit l’élimination d’eau : en
effet elle forme avec le butanol et l’acétate un hétéroazéotrope dont la Téb est relativement basse
(Téb=90,7°C). L’ester reste ainsi dans le réacteur et peut être alors purifié en fin de réaction.
IV. Rectifications successives et purification
IV-1- Rectification hétéroazéotropique
Comme cela a été vu au III, l'eau, l'acétate et le butanol donnent un hétéroazéotrope ternaire qui se
sépare en 2 couches :
l'une riche en eau (éliminée vers un doseur)
l'autre riche en acétate de butyle et en butanol (recyclée)
Cette distillation permet d’éliminer l’eau et de déplacer l’équilibre dans le sens direct tout en recyclant
le produit d’intérêt (ester).
Elimination : eau vers un
doseur
Recyclage ester + butanol : à
reflux vers le réacteur
Hétéroazéotrope ternaire issu
de la tête de colonne
Phase orga
Phase
aqueuse
Utilisation d’un florentin :
pour une décantation en
continu

Udppc PARIS de Sciences Atelier de génie chimique
ENCPB Oct 07 S. Bru, C. Canu, C. Kressmann
3
IV-2- Rectification oazéotropique
La réaction terminée, on élimine l'excès de butanol par rectification, sous forme d'azéotrope binaire
butanol – acétate (Téb=117,6°C). Cette partie est délicate car on a tendance à éliminer beaucoup
d’acétate en même temps que le butanol.
IV-3- Purification
On refroidit ensuite à température ambiante. On neutralise les traces d'acidité restante (utilisation
d’une base) et on lave l'acétate à l'eau, ensuite on dessèche l’ester par Na2CO3 anhydre. On introduit
du noir de carbone pour éliminer les impuretés. Finalement, on filtre sur adjuvant de filtration pour
éliminer le noir et le desséchant.
Ce procédé, s’il est correctement réalisé, permet d’obtenir un titre en ester dans la solution finale
proche de 99%.
V. Schéma de l’installation

Udppc PARIS de Sciences Atelier de génie chimique
ENCPB Oct 07 S. Bru, C. Canu, C. Kressmann
4
TP2 : Extraction Liquide-Liquide en continu
I. Principe de l’extraction liquide-liquide
Soit un mélange binaire constitué d’un soluté dissous dans un diluant D, que l’on doit séparer par
extraction liquide-liquide. Cette opération nécessite l’introduction d’un tiers corps liquide S appelé
solvant, non totalement miscible au mélange de base, qui extrait préférentiellement le soluté de la
solution considérée. Après contact et séparation, on obtient 2 phases :
- l’extrait : phase riche en solvant et enrichie en soluté,
- le raffinat : solution de départ épuisée en soluté.
Les deux phases à l’équilibre sont séparées par décantation. Le solvant est en général séparé du soluté
par distillation avant d’être recyclé.
II. Extraction à contre-courant dans une colonne
II-1- But et principe du TP
Le but du TP est d’étudier les caractéristiques et le fonctionnement d'une colonne garnie d’anneaux
de raschigs dans le cas d'une extraction liquide-liquide en continu, en faisant passer de l'acide
acétique (soluté) contenu dans une solution organique de Méthylisobutylcétone (MIBC : diluant) dans
de l'eau (solvant).
La phase organique légère circule de bas en haut et constitue la phase dispersée.
La phase aqueuse lourde circule de haut en bas et constitue la phase continue.
L’interphase est en haut de colonne.
Diluant
Ex : MIBC
Solvant
Ex : eau
Soluté
Ex : acide acétique
Solution homogène
Extrait (riche en solvant)
Phase lourde : Eau + acide
Raffinat (riche en diluant)
Phase légère : MIBC +
acide
acétique
Mélange composé de 2 phases non miscibles
EXTRACTION
+ DECANTATION
DISTILLATION
Diluant + Soluté
(liquide)
Diluant
Solvant + Soluté
Soluté
Solvant (liquide)
Solvant : eau
Alimentation :
MIBC + Acide acétique
Extrait : phase aqueuse
enrichie en acide
Raffinat : phase organique
appauvrie en acide
Interface
Colonne à
garnissage

Udppc PARIS de Sciences Atelier de génie chimique
ENCPB Oct 07 S. Bru, C. Canu, C. Kressmann
5
II-2- Un peu de théorie : détermination du nombre d’étages théoriques
La phase quittant le plateau 1 a la composition Ys tandis que celle entrant sur ce plateau a la
composition Xe. Donc en pied de colonne, la composition des 2 phases est donnée par le point A.
En supposant que le transfert est terminé, la phase de composition Ys sortant du plateau 1 est en
équilibre avec l’autre phase quittant ce même plateau. La composition du mélange sur le plateau 1 est
donc représentée par le point B situé à l’intersection de l’horizontale passant par A et de la courbe de
partage. En reprenant plusieurs fois le raisonnement, on aboutit à une construction en gradins de Mac
Cabe et Thiele, qui permet de définir très exactement les compositions le long de la colonne :
En pratique, le contact entre les phases se fait tout le long de la colonne sans jamais être à l’équilibre.
On définit la Hauteur Equivalente en Plateaux Théoriques (HEPT) de
la colonne par : HEPT = Z / n
où Z est la hauteur de la colonne
et n le NET issu de la construction de Mac Cabe et Thiele.
La HEPT est fonction des caractéristiques de la colonne, du mélange à
séparer et des conditions d’utilisation.
II-3- Analyses et exploitation des résultats
1. On cherche d’abord à mettre la colonne en régime permanent. Pour cela, on étudie l’évolution du
titre massique en acide de l’extrait (phase aqueuse continue) en fonction du temps.
2. Lorsque le régime permanent est atteint, on procède alors à des prises d'échantillons tout le
long de celle-ci. Les dosages permettront de déterminer les pourcentages d'acide, aux
différentes entrées et sorties de la colonne, de tracer la droite opératoire et de calculer le
nombre d'étages théoriques de la colonne, ainsi que celui de chacun des 6 éléments.On
effectuera un bilan massique sur chaque produit afin de vérifier qu’il boucle sur l’ensemble de la
colonne.
S
Ye
S
Y1 = Ys > Y2
Soluté
D
Xe
D
Xn = Xs < Xn-1
n
n - 1
Y2 > Y3
X1 < Xe
2
Yn-1 > Yn-2
X2
1
Yn > Yn-1
Xn-1
X
Y
Les compositions en acide dans le raffinat X et dans l’extrait Y
varient de bas en haut de façon continue.
On suppose que diluant et solvant sont non miscibles et que les
proportions en soluté sont faibles (D, quantité de diluant pur, et S,
quantité de solvant pur, sont constants le long de la colonne).
Bilan en soluté :
DXe + SYe = DXs + SYs sur l’ensemble de l’extracteur
Y = D/S X + Ys – D/S Xe sur la tête de colonne
Y = D/S X + Ye – D/S Xs sur le bas de colonne
ce qui donne l’équation de la droite opératoire.
Courbe d’équilibre :
Y = F(X)
Courbe opératoire : Y = G(X)
(droite si D et S constants)
Y2
Xe
X2
Xs
Ymax
X1
Ys
Y4
B
A
C
Y, teneur en soluté
dans le solvant pur
X, teneur en soluté
dans le diluant pur
D
E
Y3
X3
F
G
H
Ye
S
Yi
S
Yi+1
Soluté
D
Xi
D
Xi-1
qqs mm/s
qqs 10 cm/s
HEPT
 6
7
8
9
6
7
8
9
1
/
9
100%