DIAGNOSTIC D`UNE THALASSÉMIE Bêta thalassémie Alpha

DIAGNOSTIC D'UNE THALASSÉMIE
Bêta thalassémie
Alpha thalassémie
Les thalassémies correspondent à une diminution de synthèse des chaînes de globine. On parle de ß
thalassémie lorsque la diminution de synthèse concerne les chaînes ß, et d’a thalassémie pour les chaînes a
. Lorsque la synthèse protéique persiste, il s’agit de ß + ou a + thalassémie et en cas de disparition complète
de production de ß ° ou a ° thalassémie.
Les thalassémies sont des hémoglobinopathies mais aussi des dysérythropoïèses, avec hémolyse
intramédullaire et intravasculaire.
Les moyens diagnostics donnés ici sont fiables mais peuvent être confirmés par une électrophorèse de
l'hémoglobine, dans un laboratoire plus perfectionné (envoi de 10 ml de sang périphérique prélevé sur
EDTA).
Les thalassémies font partie des anémies hémolytiques.
BETA THALASSÉMIES
Les formes homozygotes ou maladie de Cooley :
bêta thalassémie homozygote
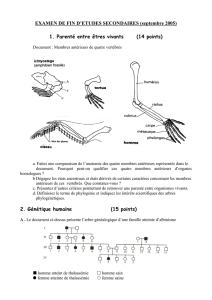
Clinique :
Elles sont caractérisées par une anémie sévère avec
ictère apparaissant dès les premiers mois, une
splénomégalie constante associée à une hépatomégalie,
un retard staturo-pondéral, une hyperplasie des os de la
face.
Diagnostic biologique :
On réalise une numération des hématies, une mesure de l'hématocrite, un dosage de l'hémoglobine et le
calcul des constantes érythrocytaires, éventuellement on numère les réticulocytes. De plus, on dose la
bilirubine et on recherche l'Hb F.
précipités d'Hb F dans une forme homozygote
L’hémogramme montre une anémie (< 70 g/l) microcytaire (VGM compris entre 60 et 65 fl) hypochrome, peu
régénérative avec présence d’hématies en cible et anisocytose. On observe aussi des corps de Howell-Jolly
et des ponctuations basophiles (se reporter aux anomalies des hématies). Les érythroblastes sont fréquents
dans le sang périphérique. L’augmentation de la bilirubine libre est le témoin de l’hémolyse.

On observe une augmentation franche de l’Hb F à la
coloration.
forme homozygote : présence d'érythroblastes.
Traitement des maladies de Cooley :
Transfusions régulières, en utilisant la desferioxamine
(Desferal) pour chélater le fer libéré par l'hémolyse
massive et éviter une hémochromatose. Ce traitement
est réalisé dans un hôpital compétent.
Les formes hétérozygotes
Elles sont cliniquement asymptomatiques.
On réalise les mêmes examens de laboratoire que pour la forme homozygote. L’hémogramme montre une
pseudo polyglobulie microcytaire. Un traitement est inutile, cependant, il faut faire attention à la descendance
pour éviter une forme homozygote et réaliser une électrophorèse des conjoints (laboratoire plus
perfectionné).
ALPHA THALASSÉMIES
frottis lors d'une alpha thalassémie
Clinique :
Il existe 4 gènes codant pour la chaîne a . La gravité de
l’expression clinique est fonction du nombre de gènes
délétés :
1- L’a ° thalassémie (a -a -,a -a -) est responsable d’un
anasarque fœto-placentaire. La délétion des quatre
gènes est létale. L’hémoglobine présente à la naissance
est l’HbG 4 ou Hb Bart.
2- (a +a -,a -a -) est responsable d’une anémie
microcytaire avec initialement une Hb Bart remplacée ensuite par une HbH(b 4).
3- (a +a +,a-a -) est responsable d’une polyglobulie microcytaire sans anémie. L’Hb Bart est inférieure à 5%
à la naissance puis disparaît.
4- (a +a +,a +a -) est silencieuse. L’Hb Bart est à l’état de trace. La délétion d’un seul gène est responsable
d'un faible trait thalassémique.
Diagnostic biologique :
Mêmes examens que pour la b thalassémie à l'exception de la recherche de l'HbF, remplacée par celle de
l'HbH. On retrouve d'autant plus d'HbH que le nombre de gènes délétés est grand. Ce diagnostic est
fortement présomptif mais demande confirmation par une électrophorèse de l'hémoglobine.
Traitement :
Le traitement repose sur la transfusion (lorsque l’hémoglobine est inférieure à 100 g/l), la lutte contre
l’infection, la prévention des carences en folates (voir anémies par carence) et de l’hémochromatose
secondaire (utilisation de desferioxamine).
1
/
2
100%