Génétique Humaine M1 Cours du 5/12/12 14h

1/7
Dystrophies héréditaires de la rétine
Génétique Humaine M1
Cours du 5/12/12 14h-16 H. Dollfus
I. Introduction : ophtalmologie et génétique
La génétique tient une place importante en ophtalmologie car plus de ¾ des malvoyances ont une
origine génétique dans nos pays développés (en Afrique la première cause est infectieuse).
Les causes génétiques peuvent toucher toutes les parties de l’œil :
antérieure = chambre antérieure avec cornée, cristallin…
postérieure = chambre postérieur avec la rétine (Objet du cours)
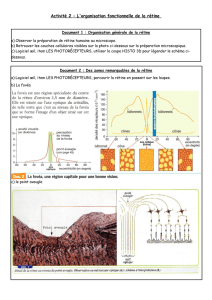
Rétine : tissu neurosensoriel du fond de d’œil
Partie centrale de la rétine : macula, pour la vision des détails, couleurs, pour la lecture.
Partie périphérique de la rétine : correspond au champ visuel, permet la vision nocturne.
La rétine est composée d’une dizaine de couches cellulaires dont les photorécepteurs : les cônes et les
bâtonnets. La lumière traverse les couches de la rétine, et permet la transduction visuelle, qui entraine
la conversion de la lumière en flux nerveux, transmis aux autres cellules jusqu’au nerf optique.
Plus précisément, la macula (zone centrale) est surtout composée de cônes, et la zone périphérique de
la rétine de bâtonnets.
II. Dystrophies héréditaires de la rétine
Elles touchent les enfants et les adultes. Il s’agit d’une dégénérescence progressive des cellules
réceptrices : cônes et bâtonnets.
Les cellules de soutien forment l’épithélium pigmentaire. Elles sont les premières touchées dans les
rétinopathies pigmentaires et entrainent la dégénérescence des autres cellules et donc de l’ensemble de
la rétine.
1. Etapes de la cascade de la transduction visuelle :
Le photon arrive sur le photorécepteur, ce qui engendre une cascade de processus biochimiques :
changement de conformation de la rhodopsine (récepteur à photon)
activation de la transducine (protéine G)
activation d’une enzyme cible : phosphodiestérase
hydrolyse GMPc
fermeture des canaux GMPc dépendants de la membrane externe du photorécepteur

2/7
La cascade aboutit à la fermeture des canaux, conduisant à une HYPERpolarisation de la membrane.
Cette hyperpolarisation est responsable de l’activation des neurones bipolaires connectés
(dépolarisation), jusqu’au cerveau.
Toutes les protéines régulatrices sont composées de sous-unités : un grand nombre de gènes codent
pour toutes ces molécules. De ce fait, une mutation d’un de ces gènes va entrainer des perturbations de
la cascade, puis un stress cellulaire, et enfin l’apoptose des cellules photoréceptrices.
Clinique :
Les dystrophies héréditaires de la rétine peuvent être divisées en 2 groupes :
- celles touchant la périphérie de la rétine = rétinopathies pigmentaires
- celles touchant le centre de la rétine = maculopathies (non traitées dans le cours)
Elles peuvent également être :
- isolées = non syndromiques
- associées à d’autres symptômes = syndromiques (ex : Syndrome d’Usher)
NB : il ne faut pas connaître les noms des pathologies par cœur, mais comprendre les mécanismes et
savoir faire le parallèle entre clinique et biologie.
2. Rétinopathies pigmentaires :
1/5000 soit 40 000 personnes atteintes en France, 1,5 millions dans le monde.
C’est une dégénérescence progressive de la vision par atteinte de l’épithélium pigmentaire et des
photorécepteurs. Elle conduit au final à la cécité (handicap sévère ++).
Rappel clinique : trépied clinique :
- hespéranopie = diminution de la vision nocturne, gène dans l’obscurité
- réduction progressive du champ visuel, pouvant parfois se restreindre au « trou de serrure » ou
« canon de fusil », très handicapant (patient marchant avec une canne blanche mais qui va sortir un livre
dans le bus car vision 10/10è, incompréhension des autres passagers du bus…)
- perte de l’acuité visuelle centrale => le handicap devient total
Examens :
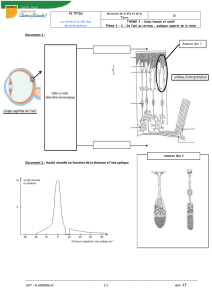
- Electrorétinogramme : mesure l’activité électrique de la rétine, teste les photorécepteurs à la
luminosité, pour classer les rétinopathies
- Fond d’œil : diminution des vaisseaux, migration pigmentée à la surface de la rétine, aspect atrophique
- Examen du champ visuel : réduit à une réponse centrale (normalement 60-70% extension)
Hétérogénéité triple du phénotype clinique :
3 niveaux clinique, génétique, moléculaire
1/ Clinique :
points noirs/points blancs sur la rétine
formes peu pigmentées
unilatéral/bilatéral
atteinte d’abord centrale puis périphérique (peu fréquent)
formes syndromiques : atteintes d’autres organes en plus

3/7
Exemples :
- Syndrome d’Usher : enfant présentant une surdité congénitale et qui va développer une cécité par
rétinopathie pigmentaire (double handicap, communication par le toucher…)
- Syndrome de Bardet-Biedl : rétinopathie pigmentaire précoce, polydactylie, obésité, atteinte rénale…
- Syndrome de Kearnes-Sayres : délétion de l’ADN mitochondrial avec ptosis (chute de la paupière),
ophtalmoplégie (paralysie de l’ œil), rétinopathie pigmentaire, troubles cardiaques, cérébrales…
2/ Génétique :
Exemple caricatural : tous les modes de transmission sont décrits :
autosomique dominant
autosomique récessif
récessif lié à l’X
dominant lié à l’X
digénique
triallélique
mitochondrial
Ces pathologies ont permis de faire des découvertes sur les modes de transmission. On a pu décrire
l’hérédité digénique (2 gènes impliqués, un allèle muté dans chaque gène), triallélique (2 allèles mutés
dans le même gène, 3è mutation dans un 2è gène), disomie uniparentale (héritage de 2 fois le
chromosome du même parent).
3/ Moléculaire :
Depuis les années 1980 : plus de 150 gènes impliqués dans la rétinopathie pigmentaire, avec des rôles
dans la fonction, la structure et le développement de la rétine, et des gènes impliqués dans la cascade.
Comme vu plus haut, les canaux sont composés de plusieurs sous-unités, et leur fermeture entraine
une hyperpolarisation.
L’arrestine est une protéine importante pour réactiver le niveau 0, récupérer l’état de repos.
Lorsque le photon va changer la conformation de la rhodopsine, elle va passer d’un état 11 cis rétinol à
all trans rétinal => jonction entre la rhodopsine et le métabolisme de la vitamine A.
Différents types de protéines impliquées :
- appartenant à la cascade de transduction (activation ou récupération), importance du métabolisme de
la vitamine A
- protéines de structure du photorécepteur
- facteurs de transcription
- protéines ubiquitaires
Rétinopathie pigmentaire autosomique dominante (RPAD) :
Premières familles étudiées, premier gène identifié : celui de la rhodopsine
Pour illustrer l’hétérogénéité : plus de 15 gènes différents impliqués

4/7
Ex : une grande famille atteinte de rétinopathie pigmentaire autosomique dominante avec des patients
moins atteints que d’autres, lié à un saut de pénétrance ou une variabilité d’expressivité.
Quand il y a une mutation de la rhodopsine : changement de conformation qui ne permet plus au
photon d’être actif.
Pour certaines familles atteintes de rétinopathie autosomique récessive (RPAR), on a pu identifier deux
mutation sur le gène de la rhodopsine => certaines mutations pour les formes AD, d’autres mutations
sur les deux allèles dans les formes AR = variabilité allélique.
Autre phénotype : Cécité nocturne congénitale stationnaire avec des difficultés en vision nocturne mais
pas de dégénérescence pigmentaire de la rétine.
D’autres gènes impliqués dans la RPAD : surtout dans les familles avec atteinte hétérogène, ce sont
des gènes ubiquitaires d’épissage des ARNm. Trois gènes sont connus : PRPC8, HPRP3, PRPF3,
exprimés partout, avec une corrélation génotype-phénotype intéressante.
La situation est la même dans la RPAR : grande hétérogénéité génétique avec atteinte d’un grand
nombre de gènes avec des fonctions différentes.
Exemple :
Amaurose congénitale de Leber : forme très précoce de rétinopathie pigmentaire, les enfants
naissent avec une rétine quasiment non fonctionnelle, elle dégénère très rapidement. 17 gènes
sont impliqués sur le mode récessif.
Quelques gènes :
- GUCY2D : enzyme guanylate cyclase, spécifique de la cascade de transduction
- RPE65 ++ : importante pour le cycle du dérivé de la vitamine A, premier gène sur lequel on
a fait des essais de thérapie génique, sert à la régénération des dérivés de la vitamine A au
niveau de l’épithélium pigmentaire.
- RPGRIP1 : impliqué dans le transport du photorécepteur, pour le transport des protéines
du segment interne vers le segment externe du photorécepteur (cil connecteur), n’agit pas
au niveau du cycle ni de la cascade.
- CRX : facteur de transcription, gène impliqué dans le développement : il va allumer
d’autres gènes importants pour le développement, à partir de précurseurs des
photorécepteurs.
=> hétérogénéité des gènes impliqués avec des rôles très différents au niveau du photorécepteur.
Les photorécepteurs sont composés de disques empilés les uns sur les autres, formés en majorité
par la rhodopsine et protéines de stabilisation RDS-ROM1 pour que la structure soit maintenue.
Modèle du chien Briard atteint de rétinopathie pigmentaire délétée dans RPE65 : on a pu testé les
premières étapes de thérapie génique dans un œil et pas dans l’autre => le chien voyait avec l’œil
traité.

5/7
Problème du conseil génétique :
Mariage entre deux personnes atteintes d’amaurose congénitale de Leber : quel risque pour la
descendance ?
C’est une maladie autosomique récessive, il faut savoir si les patients sont mutés dans lequel des 17
gènes.
Si les deux personnes sont homozygotes pour un même gène => 100% de leurs enfants atteints
Si elles sont homozygotes pour les gènes différents => risque de 0% car l’enfant reçoit un seul allèle
de chaque gène.
NB : Mucoviscidose : un seul gène, plus de 1000 mutations.
Il faut tester le patient pour rechercher la mutation, et ensuite on fait le conseil génétique.
Quand un couple de parents a un enfant atteint d’amaurose congénitale de Leber => 25% de risque
d’avoir un 2è enfant atteint. On recherche la mutation dans le gène pour avoir accès au DPN ou DPI.
Variabilité allélique :
Exemple : gène ABCR = ABC A4
Sur le mode récessif, les mutations du gène conduisent à la rétinopathie pigmentaire. D’autres
mutations dans ce gène peuvent conduire à une maculopathie : Maladie de Stargardt.
=> mutation différente dans un même gène conduisant à un phénotype différent, pourquoi ?
ABC A4 a un rôle spécifique dans la cascade de la transduction visuelle, il agit sur un dérivé de la
vitamine A : rôle de « flippase » sur le dérivé activé de la rhodopsine (all-trans-retinal).
Quand 1 mutation faux sens : facteur de risque de DMLA (dégénérescence maculaire liée à
l’âge)
Quand 2 mutations faux sens : maladie de Stargardt (niveau intermédiaire)
Quand 2 mutations tronquantes (pas de protéine produite) : rétinopathie pigmentaire
Les « fausses récessives » :
Digénisme : mutation hétérozygote dans RDS et mutation hétérozygote dans ROM1, agit sur
l’interaction entre ces 2 protéines in vivo. Risque vertical de 25% : le parent atteint transmet les 2
mutations à l’enfant => piège en génétique.
On va découvrir des situations identiques avec le séquençage haut débit. Phénomène décrit aussi
pour la surdité.
Rétinopathie pigmentaire liée à l’X :
Une des formes les plus sévères, 13-15% des patients. Les femmes conductrices présentent des
signes cliniques. Deux gènes identifiés, localisés au niveau du cil connecteur.
Rétinopathie pigmentaire syndromique :
2 types :
1/ Syndromes sensoriels => ciliopathies (voir cours correspondant)
2/ Syndromes « House-keeping » ou maladies générales de la maintenance cellulaire :
Toutes ces pathologies portent le nom de ceux qui les ont décrites (d’un point de vue clinique),
mais en fait il y a des syndromes qui se chevauchent, d’où la nécessité d’essayer de faire une
classification plus biologiques de ces syndromes.
Si on comprend les mécanismes biologiques, on peut mieux trouver un traitement.
 6
7
6
7
1
/
7
100%