ÉTUDE des étapes précoces du cycle DE réplicatiON du virus d

GAËL VIDRICAIRE
ÉTUDE DES ÉTAPES PRÉCOCES
DU CYCLE DE RÉPLICATION DU VIRUS
D’IMMUNODÉFICIENCE HUMAINE DE TYPE 1
DANS LES CELLULES TROPHOBLASTIQUES :
Vers une compréhension de la transmission materno-fœtale
Thèse présentée
à la Faculté des études supérieures de l’Université Laval
dans le cadre du programme de doctorat en microbiologie-immunologie
pour l’obtention du grade de Philosophiæ doctor (Ph.D.)
BIOLOGIE MÉDICALE
FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL
QUÉBEC
2006
© Gaël Vidricaire, 2006

ii
Résumé
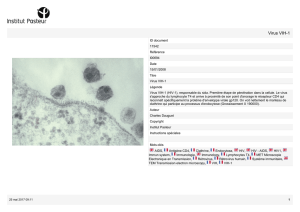
Plus de deux millions d’enfants dans le monde vivent actuellement avec le virus
d’immunodéficience humaine de type 1 (le VIH-1). De plus, les femmes, particulièrement
celles en âge de procréer, sont plus vulnérables à cette infection. Or, 90% des infections par
ce rétrovirus chez l’enfant sont attribuables à la transmission de la mère à l’enfant (TME).
Quoique des traitements antirétroviraux soient disponibles pour la prévenir, seulement une
infime proportion des femmes séropositives y a accès encore aujourd’hui. On estime donc
que la transmission verticale du VIH-1 est un problème de santé public alarmant, non
seulement pour les générations d’aujourd’hui, mais aussi pour celles de demain.
La contamination fœtale est l’une des voies par laquelle la TME peut se produire.
Cependant, les mécanismes qui y sont associés demeurent peu connus, particulièrement en
ce qui concerne l’infection directe des trophoblastes, éléments structuraux du placenta. Afin
d’éclaircir ce sujet, nous avons étudié, dans cette thèse, les évènements précoces du cycle
réplicatif du VIH-1 dans les trophoblastes, première étape conduisant à l’infection d’une
cellule cible.
Nous avons découvert que le mécanisme d’infection des trophoblastes est inhabituel pour
ce rétrovirus. En effet, le VIH-1, au contact de ces cellules, est internalisé par celles-ci et se
retrouve alors dans les endosomes. Nous avons établi que l’endocytose du VIH-1 se fait par
une voie indépendante de la clathrine, des cavéoles et de la dynamine-2, mais requérant
toutefois que le cholestérol membranaire soit libre. Nous avons suivi le parcours
intracellulaire des particules virales et noté qu’elles se rendaient principalement aux
endosomes tardifs, transit contrôlé par les protéines Rab5 et Rab7. Quoique ce transport
entraîne la dégradation d’une grande partie des virions, il est de façon étonnante essentiel à
leur processus infectieux dans les trophoblastes. Finalement, l’accès du VIH-1 au
cytoplasme cellulaire se fait en l’absence des protéines de l’enveloppe virale, la gp120 et la
gp41, suggérant que le virus fusionne dans les endosomes grâce à la machinerie cellulaire
présente. Les données présentées dans cette thèse décrivent donc une nouvelle voie
d’infection pour le VIH-1. La compréhension de ce processus est essentielle si l’on veut
mieux contrôler un jour la transmission mère-enfant du VIH-1 durant la grossesse et/ou
trouver des solutions alternatives aux antirétroviraux existants.

iii
Abstract
More than two million children under fifteen years of age are currently living with the
human immunodeficiency virus type 1 (HIV-1) worldwide and 90% of these infections are
associated with mother-to-child transmission (MTCT) of this retrovirus. Women,
particularly those of child-bearing age, are highly susceptible to HIV-1 infection. In spite of
available antiretroviral treatments to prevent MTCT, only a minority of infected women
have access to these treatments. Hence, vertical transmission of HIV-1 is an alarming
public health issue for both current and future generations.
One of the postulated models for how HIV-1 is transmitted by the mother is foetal
contamination. However, the mechanisms underlying such an event are poorly understood.
In particular, the process whereby HIV-1 may directly infect trophoblasts, the structural
cells of the placenta, is unknown. In this thesis, we have studied the early events associated
with HIV-1 life cycle in trophoblasts, the first step towards infecting a target cell.
Our data demonstrate that the mechanism whereby HIV-1 infects trophoblasts is unusual
for this retrovirus. Upon contact with these cells, HIV-1 is rapidly and massively
endocytosed. We have tracked the step-by-step movements of incoming particles and found
that HIV-1 traffics primarily towards late endosomes, via Rab5 and Rab7. Surprisingly,
although this transit leads to the degradation of the majority of the internalized virions, it is
necessary for HIV-1 to establish a productive infection in these cells. In addition, we found
that endocytosis of HIV-1 in these placental cells relies on a clathrin-, caveolae- and
dynamin-independent pathway that requires free membrane cholesterol. Finally, viral entry
occurs in the absence of the viral envelope glycoproteins, gp120 and gp41, suggesting that
HIV-1 undergoes fusion within the endosomes via the host cell machinery.
Collectively, the data presented in this thesis describe a novel infection pathway for HIV-1.
An understanding of this unique process is essential if we are to learn how to control
MTCT and/or find alternate solutions to existing antiretroviral drugs.

iv
Avant-propos
Le sujet de recherche qu’on m’a proposé au début de mes études doctorales m’a tout de
suite plu. La transmission verticale du VIH-1 est une question qui m’interpellait à la fois
comme femme, mère et chercheure. De plus, il s’agissait d’un nouveau projet pour le
laboratoire, donc tout était à faire. Ceci comportait un certain risque mais également un
beau défi.
Je n’aurais pu accomplir ce travail de recherche seule et je tiens à remercier plusieurs
personnes qui m’ont grandement épaulée. D’abord, mon directeur de thèse, le Dr Michel J.
Tremblay, qui a pris le risque de m’accepter dans son laboratoire alors que je n’avais pas
poursuivi un trajet académique régulier. La rigueur scientifique et les très grandes qualités
de gestionnaire du Dr Tremblay en font un patron exceptionnel. Je tiens aussi à souligner sa
générosité et sa très grande disponibilité. De plus, l’immense liberté qu’il m’a donnée et la
confiance qu’il m’a accordée m’ont été fort précieuses. Bref, j’ai eu la chance d’évoluer
dans un environnement scientifique incomparable.
Je me dois de remercier les Dr Benoît Barbeau et Sachiko Sato pour leurs suggestions, leurs
commentaires et leurs conseils judicieux, tant au plan technique et scientifique que
personnel. Je souligne également l’aide offerte par tous les membres de l’équipe du Dr
Tremblay, dont certains ont déjà quitté son laboratoire, mais avec qui j’ai beaucoup discuté
tout au long de mes études.
Je voudrais aussi souligner toute l’affection et le support moral que m’a prodigués toute ma
famille, particulièrement mon mari, Serge Leclerc, mes enfants, Arnaud et Alexia sans qui
je n’aurais pas su y arriver.
Finalement, je remercie sincèrement les Instituts de recherche en santé du Canada (IRSC)
pour leur support financier tout au long de mes études.

v
Je présente une thèse avec insertion d’articles. Cette thèse inclut les cinq articles suivants :
i) Vidricaire, G., Tardif, M.R., Tremblay, M.J. The low viral production in
trophoblastic cells is due to a high endocytic internalization of the human
immunodeficiency virus type 1 and can be overcome by the pro-inflammatory
cytokines tumor necrosis factor-α and interleukin-1. (2003) The Journal of
Biological Chemistry; Vol. 278, No. 18, pp. 15832–15841.
ii) Vidricaire, G., Gauthier, S., Tremblay, M.J. HIV-1 infection of human trophoblasts
is independent of gp120/CD4 interactions but relies on heparan sulfate
proteoglycans. (2005). Cet article a été soumis pour publication à la revue
scientifique Journal of Virology.
iii) Vidricaire, G., Imbeault, M., Tremblay, M.J. Endocytic host cell machinery plays a
dominant role in intracellular trafficking of incoming human immunodeficiency
virus type 1 in human placental trophoblasts. (2004) Journal of Virology;
78(21):11904-15.
iv) Vidricaire, G., Tremblay, M.J. HIV-1 infection of human polarized trophoblasts is
associated with internalization via clathrin/caveolae-independent endocytosis and
free cholesterol. (2005) Cet article a été soumis pour publication à la revue
scientifique Traffic.
v) Vidricaire, G., Tremblay, M.J. Rab5 and Rab7 but not ARF6 govern the early
events of HIV-1 infection in polarized human placental cells. (2005). Journal of
Immunology; 175(10):6517-6530.
Gaël Vidricaire est l’auteure principale de tous ces articles. Les résultats présentés à la
figure 2 de l’article 1 ont été obtenus par Mélanie Tardif, alors que ceux présentés à la
figure 6B de l’article 2 ont été générés par Sonia Gauthier. De plus, pour l’article 3,
Michael Imbeault a effectué la programmation permettant l’analyse des images obtenues
par microscopie confocale par le logiciel Metamorph et a contribué à produire les images
par le logiciel Photoshop. Outre ces trois cas, le design expérimental, l’ensemble des
expériences et la rédaction scientifique de chacun des articles ont été réalisés en totalité par
l’auteure de cette thèse. Michel J. Tremblay en a assuré la supervision scientifique.
Ces cinq articles présentés dans cette thèse correspondent de façon intégrale aux articles qui
ont été soumis pour publication ou, le cas échéant, qui sont déjà publiés.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
452
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
452
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
1
/
469
100%






![Poster CIMNA journée CHOISIR [PPT - 8 Mo ]](http://s1.studylibfr.com/store/data/003496163_1-211ccc570e9e2c72f5d6b6c5d46b9530-300x300.png)



