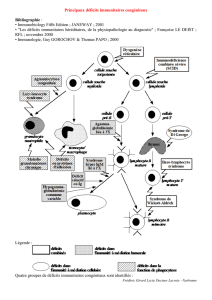
Mécanismes des anomalies du développement du système

Galudec Pierre-Marie, Lacomblet Lucie
24/10/12, UE Immunopathologie,
Mécanismes des anomalies du développement du système immunitaire
Pr Virginie Gandemer , Dr Cédric Ménard
Diapo dispo sur le réseau pédagogique
Mécanismes des anomalies du
développement du système immunitaire
Introduction
Plus de 200 déficits immunitaires héréditaires. Ce sont des maladies génétiques rares (1 naissance
sur 5000).
Les différents déficits immunitaires (DI) sont classés en fonction du défaut immunologique:
dans l'ordre de fréquence :
–DI humoral isolé (moins graves, plusieurs possibilités de traitement) : 60%
–DI combinés cellulaire/humoral (touche T et B) : 20%
–DI des cellules phagocytaires : 10%
–DI du complément : 1%
–Autres DI innés
–DI de l’homéostasie du système immunitaire : touche la régulation de la réponse
immunitaire dans son ensemble.
–DI complexes et/ou syndromiques
Les DI prédisposent aux infections +++ et/ou cancers et/ou auto-immunité.
Isolément, ces différents DI sont tous très rares.
I- Définition moléculaire des déficits primitifs
La plupart des anomalies moléculaires ont été identifiées et ont permis de mieux comprendre
la physiopathologie.
Les analyses moléculaires peuvent:
• Fournir le diagnostic de certitude
• Aider au conseil génétique : permettant le diagnostic anténatal précoce et l’identification
des porteurs .
• Établir un diagnostic dans des présentations atypiques
• Donner des informations pronostiques pour les déficits où une forte corrélation
géno/phénotype existe
1/25

• Permettre l ’ identification présymptomatique des individus atteints de formes graves :
conduisant à des interventions lourdes précoces (greffe de CSH)
Mais un gène ne code pas pour 1 protéine : 22000 gènes codent 100.000 protéines (épigénétique :
épissage alternatif, régulation post transcriptionnelle…) .
Bon nombre de DICV (déficit immunitaire commun variable) n’ont pas de causes moléculaires
identifiées comme la plupart des patients ayant un déficit en sous classes d’Ig .
Une même mutation peut être à l'origine de plusieurs maladies différentes.
Une même maladie peut toucher plusieurs gènes.
2/25

II- Données épidémiologiques
Les DI concernent1/5000 naissances vivantes .
Les données épidémiologiques françaises accessibles sur le site du CEREDIH (Centre de Référence
pour les Déficits Immunitaires Héréditaires): réseau clinico-biologique spécialisé dans la prise en
charge des DIH. D'où l'importance du rescensement des patients atteints.
La prévalence en France en 2012 est de 5,22/105 habitants , soit 3342 patients vivants.
III- Déficit de l’immunité humorale
Ce sont les plus fréquents parmis les DI.
On la définie par une anomalie entrainant un défaut de synthèse d'Ig.
Ils peuvent être secondaires à une anomalie des ly. T (plus de « help » par les T CD4) .
En fonction du sous-type d'IgG déficitaire, on aura telle ou telle prédisposition à certains types
d’infections.
A- Agammaglobulinémie
Les gènes mutés sont impliqués dans l’ontogénie des lymphocytes B . Cela implique l'absence de
lymphocytes B circulants dans le sang, et donc l'absence de synthèse d’Ig .
Les premières manifestations cliniques apparaissent à partir de l'âge de 6 mois (1ères années de
vie) .
On peut observer des infections récurrentes (bactériennes ++, giardiase, entérovirus) .
Il existe deux types principaux d'agammaglobulinémie :
- L'agammaglobulinémie de Bruton ou agammaglobulinémie liée à l’X (85% des cas)
–Récessif
–Fréquence : 1/250000
–Mutation du gène BTK codant pour la Bruton tyrosine kinase impliquée dans la
signalisation du BCR et du pré-BCR .
–Blocage des Ly B au stade pré-B.
–Absence de Ly B circulants, absence de production d’Ig.
Les enfants atteints présentent des infections banales à répétition comme une rhinopharyngite, une
bronchite, etc. On peut parler de “signe du carnet de santé” car leurs carnets sont très fournis.
3/25

- L'agammaglobulinémie par transmission autosomique récessive (15% des cas)
–Mutation de la chaîne lourde Cm
–Mutation de la pseudo-chaîne légère l5
–Mutation de CD79a
–Mutation de la protéine adaptatrice BLNK (=B cell linker protein) : tableau
identique au Bruton
Implique le blocage à différents stades de différenciation B .
Traitement : substitution à vie par Ig polyvalentes.
B- Cas clinique
Gwendal
ATCD familiaux : sans particularité
ATCD perso : Infection urinaire néonatale à E.Coli (RVU)
Anamnèse :
Vaccinations sans complication .
Bronchites et OMA à répétition dès les premiers mois de vie.
Pas altération de la croissance staturo pondérale, ni du développement psychomoteur.
À 2 ans : pneumonie franche lobaire aigue d’évolution rapidement favorable sous bi antibiothérapie
(claforan ,nétromycine).
4/25

Résultats :
Recherche d’un reflux gastro-oesophagien
Recherche d’une mucoviscidose
NFS normale
Dosage des immunoglobulines : hypogammaG . Ig G < 1.8 g/l, Ig A < 0.3 g/ l, IgM < 0.2 g/l,
Réponse vaccinale :
–anticorps anti diphtériques <0.025 UI/ml (non protecteurs )
–anticorps antitétaniques = 0.125 UI/ml (limite inférieure)
–mauvaise réponse vaccinale.
=> Il a peu d'Ac et en plus il ne pas mettre en place une bonne mémoire immunitaire.
Rappel : Répartition des Ig sériques chez le nouveau-né et l’enfant :
Le taux d'IgG normal est acquis à l'âge de 6 ans.
Celui des IgA est atteint à la puberté.
Le taux d'IgM atteint un plateau vers l'âge de 1 an.
Passage transplacentaire des IgG à partir du 3ème trimestre de grossesse :
–Attention aux bébés prématurés !
–Toutes les sous-classes (IgG1 et IgG3++)
–Immunité passive de 0 à 6 mois de vie
Etude lymphocytaire :
Population lymphocytaire : Lymphocytes B :19 %
Lymphocyte T : 48 % dont CD4 : 23% et CD8 : 35 %
Test de prolifération lymphoblastique : réactivité normale aux antigènes de rappel et réactivité
normale aux mitogènes sauf avec le PWM.
Diagnostic :
Hypogammaglobulinémie avec présence de lymphocytes B circulants.
==> Déficit commun variable
5/25
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
1
/
25
100%