Effet du 5-aza-2`-désoxycytidine et de l`acide valproïque sur l

0
UNIVERSITÉ DE NANTES
FACULTÉ DE PHARMACIE
ANNÉE 2008 N°61
THÈSE
pour le
DIPLÔME D’ÉTAT
DE DOCTEUR EN PHARMACIE
PAR
Thomas DAVID
Présentée et soutenue publiquement le 4 Décembre 2008
Effet du 5-aza-2’-désoxycytidine et de l’acide
valproïque sur l’expression d’antigènes
tumoraux dans le mésothéliome pleural malin
Président : M. Jacques AUBRY, Professeur d’Immunologie
Membres du jury : Mlle. Marianne LE PRIELLEC, Docteur en
Pharmacie
M. Stéphane BIRKLE, Maître de Conférences en
Immunologie

1
Sommaire
LISTE DES FIGURES ................................................................................................................................... 5
LISTE DES TABLEAUX ............................................................................................................................... 7
ABREVIATIONS ......................................................................................................................................... 8
Première partie : introduction générale ............................................................................................... 11
I. Le mésothéliome pleural malin .......................................................................................................... 12
I. 1. Définition et situation anatomique ........................................................................................... 12
I. 2. Données épidémiologiques ....................................................................................................... 13
I. 3. Facteurs étiologiques et mécanismes pathogéniques .............................................................. 14
I.3.1. Les fibres minérales .............................................................................................................. 14
I.3.2. Le virus SV40 ......................................................................................................................... 16
I.3.3. Autres facteurs étiologiques ................................................................................................. 17
I.3.4. Mécanismes moléculaires .................................................................................................... 18
I. 4. Etablissement du diagnostic ...................................................................................................... 20
I.4.1. Analyses immunocytochimiques .......................................................................................... 21
I.4.2. Analyses immunohistochimiques ......................................................................................... 21
I.4.3. Dosage de marqueurs solubles ............................................................................................. 23
I. 5. Modalités thérapeutiques ......................................................................................................... 26
I.5.1. La chirurgie ........................................................................................................................... 26
I.5.2. La chimiothérapie ................................................................................................................. 27
I.5.3. La radiothérapie .................................................................................................................... 28
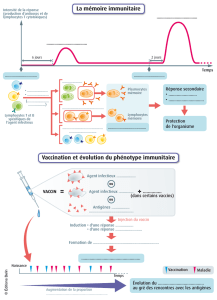
II. L'immunité anti-tumorale ................................................................................................................. 28
II.1. Emergence d'une théorie : de l'immunosurveillance à l'immunoédition ................................. 28
II.2. Les effecteurs de la surveillance immunitaire anti-tumorale.................................................... 31
II.2.1. Les lymphocytes T cytotoxiques .......................................................................................... 31
II.2.2. Les cellules dendritiques ...................................................................................................... 35
II.2.3. Les autres cellules effectrices de l'immunité anti-tumorales .............................................. 46

2
II.3. Les antigènes de tumeurs .......................................................................................................... 51
II.3.1. Les différentes techniques d'identification d'antigènes de tumeurs .................................. 51
II.3.2. Classification des antigènes ................................................................................................. 53
II.4. Les mécanismes d'échappement à la réponse antitumorale .................................................... 58
II.4.1. Phénomène de tolérance. ................................................................................................... 59
II.4.2. Influence des facteurs solubles sécrétés par les cellules tumorales. .................................. 60
II.4.3. Défaut d’activation des lymphocytes T ................................................................................ 60
II.4.4. Résistance à l’apoptose ....................................................................................................... 61
II.4.5. Lymphocytes T régulateurs. ................................................................................................. 61
II.4.6. Localisation des tumeurs. .................................................................................................... 62
III. Immunothérapie du mésothéliome ................................................................................................. 62
IV. Intérêt de l’utilisation de molécules hypométhylant l’ADN et d’inhibiteurs d’histone
désacétylase .......................................................................................................................................... 63
IV. 1. Rôle et mécanisme de la méthylation de l’ADN et de l’acétylation des histones .................. 63
IV.1.1. La méthylation de l’ADN ..................................................................................................... 63
IV.1 2. L’acétylation des histones .................................................................................................. 65
IV.2. Modifications épigénétiques et oncogenèse ........................................................................... 66
IV.3. Mécanisme d’action du 5-azacytidine, du 5-aza-2’-désoxycytidine et de l’acide valproïque . 66
IV.3.1. Le 5-aza-2’-désoxycytidine et du 5-azacytidine ................................................................. 66
IV.3.2. L’acide valproïque .............................................................................................................. 68
IV. 4. Méthylation de l’ADN, acétylation des histones et expression d’antigènes tumoraux. ......... 68
IV.4.1. Généralités ......................................................................................................................... 68
IV.4.2. Molécules hypométhylantes et expression des antigènes tumoraux ................................ 70
V. Choix des antigènes tumoraux étudiés ............................................................................................. 71
V.1. Les antigènes tumoraux de type testiculaire ........................................................................... 71
V.2. La mésothéline et la mucine-1 .................................................................................................. 72
VI. Objectif de l’étude ............................................................................................................................ 73
Deuxième partie : partie expérimentale ............................................................................................... 74

3
MATERIEL ET METHODES ...................................................................................................................... 75
I. Patients ............................................................................................................................................... 75
II. Lignées cellulaires .............................................................................................................................. 75
III.Traitement des cellules ..................................................................................................................... 75
III.1. Préparation des solutions-mères ............................................................................................. 75
III.2. Comparaison de l’efficacité du 5-azacytidine et du 5-aza-2’-désoxycytidine sur l’induction
de l’expression d’antigènes tumoraux ............................................................................................. 75
III.3. Choix du mode de traitement .................................................................................................. 76
III.4. Test effet-dose du 5-aza-2’-désoxycytidine et de l’acide valproïque ...................................... 76
IV. Extraction des ARN totaux ............................................................................................................... 77
V. Traitement à la DNase et transcription inverse (RT) ......................................................................... 77
VI. Réaction d’amplification génique (PCR) ........................................................................................... 78
VII. PCR en temps-réel ........................................................................................................................... 79
VIII. Etude de la persistance d’expression des gènes induits par le traitement au 5-azaCdR ............... 80
IX. Fixation de l’anticorps anti-NY-ESO-1 .............................................................................................. 80
X. Activation de la réponse immunitaire. .............................................................................................. 80
X.I. Etude de la réponse immunitaire après traitement des cellules avec 5-azaCdR ou VPA .......... 80
X.II. Traitement des cellules avec 5-azaCdR et VPA ......................................................................... 81
RESULTATS ............................................................................................................................................. 82
I. Vérification de l’intégrité des ARN ..................................................................................................... 82
II. Comparaison de l’efficacité du 5-azaCR et du 5-azaCdR sur l’induction de l’expression
d’antigènes tumoraux (CTA et antigènes de différenciation) ............................................................... 82
III. Choix du mode de traitement .......................................................................................................... 83
IV. Test effet-dose du 5-azaCdR et de l’acide valproïque ..................................................................... 84
IV.1. Etude de la viabilité cellulaire .................................................................................................. 84
IV.2. Etude de la prolifération cellulaire........................................................................................... 85
IV.2.1. Effets du 5-azaCdR .............................................................................................................. 85
IV.2.2. Effets du VPA ...................................................................................................................... 86

4
V. PCR quantitative en temps réel......................................................................................................... 86
V.1. Mise au point de la qPCR .......................................................................................................... 86
V.2. Induction de l’expression du transcrit NY-ESO-1 ...................................................................... 87
VI. Induction de l’expression de différents antigènes de tumeur par le 5-azaCdR et le VPA. .............. 90
VII. Etude de la persistance d’expression des gènes induits par le traitement au 5-azaCdR ................ 91
VIII. Fixation de l’anticorps anti-NY-ESO-1 ............................................................................................ 91
IX. Activation de la réponse immunitaire .............................................................................................. 92
IX.1. Traitement des cellules avec 5-azaCdR ou VPA ....................................................................... 92
IX.2. Traitement des cellules avec combinaison de 5-azaCdR et de VPA ......................................... 94
DISCUSSION ........................................................................................................................................... 97
PERSPECTIVES ...................................................................................................................................... 102
CONCLUSION ....................................................................................................................................... 103
BIBLIOGRAPHIE .................................................................................................................................... 104
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
1
/
131
100%