Biologie moléculaire

Référentiel régional
Biologie moléculaire
Version 4 – 2016 / 2017

Table des matières
Chapitre 1 - Introduction ________________________________________________________ 6
1.1. Présentation du référentiel _______________________________________________________ 7
1.2. Groupe de travail biologie moléculaire – Actualisation 2016 _____________________________ 8
Chapitre 2 - Généralités sur les méthodes d’analyse des anomalies génomiques ___________ 9
2.1. Les anomalies de structures chromosomiques : les translocations _______________________ 10
2.1.1. Méthodes par extraction des acides nucléiques ___________________________________________ 10
2.1.2. Méthodes in situ ____________________________________________________________________ 10
2.2. Amplification et délétion génique _________________________________________________ 11
2.2.1. Méthodes par extraction des acides nucléiques ___________________________________________ 11
2.2.2. Méthodes in situ ____________________________________________________________________ 11
2.3. Les anomalies des gènes : les mutations ____________________________________________ 12
2.3.1. Méthodes par extraction des acides nucléiques ___________________________________________ 12
2.3.2. Méthodes in situ ____________________________________________________________________ 12
2.4. Les nouvelles méthodes d’analyse – Nouveautés 2016 ________________________________ 12
2.4.1. L’analyse par séquençage haut débit ____________________________________________________ 12
2.4.2. L’analyse par ADN tumoral circulant ou biopsie liquide _____________________________________ 13
Chapitre 3 - Prescription en pratique - Actualisation 2016 ___________________________ 14
Chapitre 4 - Essais de therapie ciblée en lien avec la plateforme de génétique des tumeurs _ 17
4.1. Programme de détection prospective des biomarqueurs émergents _____________________ 18
4.2. Programme AcSé - Actualisation 2016 _____________________________________________ 18
Chapitre 5 - Gynécologie – Actualisation 2016 _____________________________________ 20
5.1. Infections HPV _________________________________________________________________ 21
5.2. Cancer de l’endomètre et syndrome de lynch ________________________________________ 22
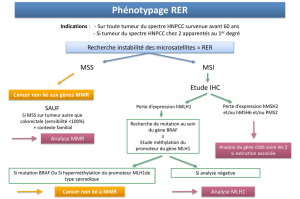
5.2.1. Cancer de l’endomètre et syndrome de Lynch : test MSI /méthylation promoteur MLH _______ 22
5.2.2. Tableau de synthèse du test ___________________________________________________________ 24
5.3. Sarcome du stroma endométrial de bas grade _______________________________________ 25
5.4. Carcinome de l’ovaire, de la trompe, du péritoine, de haut grade _______________________ 26
5.5. Tumeur de la granulosa et mutation FOXL2 _________________________________________ 27
Chapitre 6 - Hématologie ______________________________________________________ 28
6.1. Syndromes myéloproliferatifs ____________________________________________________ 34
6.1.1. Leucémie myéloïde chronique (LMC) ____________________________________________________ 34
6.1.2. La polyglobulie primitive (PV) ou maladie de Vaquez (MV) __________________________________ 36
6.1.3. La Thrombocytémie Essentielle (TE) ____________________________________________________ 37
6.1.4. La myélofibrose primitive (MF) ou splénomégalie myéloïde chronique _________________________ 38
6.2. leucémies aigues _______________________________________________________________ 38
6.2.1. Leucémies aiguës myéloïdes (LAM) _____________________________________________________ 38

6.2.2. Leucémies aiguës lymphoblastiques (LAL) ________________________________________________ 40
6.3. Syndromes lymphoprolifératifs ___________________________________________________ 41
6.3.1. La leucémie lymphoïde chronique (LLC) _________________________________________________ 41
6.3.2. Les lymphomes _____________________________________________________________________ 41
6.3.3. Le myélome multiple (MM) – Actualisation 2016 __________________________________________ 45
Chapitre 7 - Mélanome ________________________________________________________ 46
7.1. mélanome métastatique ________________________________________________________ 47
7.1.1. Thérapie ciblée des mélanomes métastatiques : génotypages BRAF/NRAS/KIT __________________ 47
7.1.2. Tableaux de synthèse des tests ________________________________________________________ 49
Chapitre 8 - Neurologie ________________________________________________________ 52
8.1. génotypage utile à visée thérapeutique, diagnostique et pronostique ____________________ 53
8.1.1. La recherche de la co-délétion 1p19q à visée thérapeutique _________________________________ 53
8.1.2. La recherche de la co-délétion 1p19q à visée diagnostique et pronostique _____________________ 53
8.1.3. Génotypage utile à visée pronostique : méthylation du géne MGMT __________________________ 55
8.1.4. Autres examens utiles _______________________________________________________________ 56
Chapitre 9 - orl _______________________________________________________________ 57
Chapitre 10 - Pathologie digestive ________________________________________________ 58
10.1. Cancer colorectal ____________________________________________________________ 59
10.1.1. Diagnostic du syndrome de Lynch : test MSI /génotypage BRAF/méthylation promoteur MLH1 ____ 59
10.1.2. Traitement adjuvant des cancers colorectaux de stade II : test MSI ___________________________ 61
10.1.3. Thérapie ciblée des cancers colorectaux métastatiques : génotypage RAS/BRAF _________________ 62
10.1.4. Programme INCa de détection prospective des biomarqueurs émergents ___________________ 63
10.1.5. Programme AcSé ____________________________________________________________________ 63
10.1.6. Polypose __________________________________________________________________________ 64
10.1.7. Tableaux de synthèse des tests ________________________________________________________ 65
10.2. Gist _______________________________________________________________________ 70
10.2.1 Génotypage KIT /PDGFRA à visée diagnostique ___________________________________________ 70
10.2.2 Génotypage KIT/ PDGFRA et traitement par imatinib _______________________________________ 70
10.2.3 Tableaux de synthèse des tests ________________________________________________________ 71
10.3. Cancers gastriques ___________________________________________________________ 72
10.3.1. A visée thérapeutique ________________________________________________________________ 72
10.3.2. Programme AcSé ____________________________________________________________________ 73
10.3.3. Tableau des tests ___________________________________________________________________ 74
10.4. Cancer œsophage, canal anal ___________________________________________________ 75
10.5. Cancer du foie _______________________________________________________________ 75
10.3.4. Cholangiocarcinome et le programme AcSé ______________________________________________ 75
10.3.5. Cancer du foie et le programme AcSé ___________________________________________________ 75
10.6. Tumeur bénigne du foie _______________________________________________________ 75
10.3.6. Génotypages utiles à la classification des adénomes hépatocellulaires_________________________ 76
10.7. Cancer du pancréas ___________________________________________________________ 76
10.8. Tumeurs endocrines __________________________________________________________ 76

10.9. Lymphomes digestifs _________________________________________________________ 76
10.10. Sarcomes (Autres que Gist) ____________________________________________________ 76
Chapitre 11 - Pneumologie ______________________________________________________ 77
11.1. A visée thérapeutique ________________________________________________________ 78
11.1.1. Prescription d’inhibiteurs de tyrosine kinase d’EGFR _______________________________________ 78
11.2. Prescription d’inhibiteurs de ALK et MET : Crizotinib (Xalkori) ________________________ 80
11.3. A visée diagnostique et pronostique _____________________________________________ 82
11.4. Les programmes en cours ______________________________________________________ 82
11.4.1. Programme de détection prospective des biomarqueurs émergents dans le cancer du poumon ____ 82
11.4.2. Programme AcSé ____________________________________________________________________ 85
Chapitre 12 - Sarcome - Actualisation 2016 ________________________________________ 87
12.1. Tableaux de synthèse des tests réalisés sur la plateforme de génétique des cancers de Midi-
Pyrénées 88
12.1.1. Réarrangement du gène SYT __________________________________________________________ 88
12.1.2. Réarrangement du gène EWSR1 (EWS) __________________________________________________ 89
12.1.3. Réarrangement du gène DDIT3 ________________________________________________________ 91
12.1.4. Réarrangement du gène ALK __________________________________________________________ 92
12.1.5. Réarrangement du gène TFE3 _________________________________________________________ 93
12.1.6. Réarrangement du gène ETV6 _________________________________________________________ 94
12.1.7. Amplification du gène MDM2 _________________________________________________________ 95
12.1.8. Translocation t(17 ;22)(q22 ; q23) ______________________________________________________ 96
12.1.9. Amplification de c-myc _______________________________________________________________ 97
12.1.10. Sarcomes du stroma endométrial ______________________________________________________ 98
12.1.11. Mutation du gène de la béta-caténine (CTNNB1) __________________________________________ 99
12.1.12. Génotypage KIT / PDGFRA ___________________________________________________________ 100
12.2. Tableaux de synthèse des tests réalisés à l’Institut Bergonié _________________________ 101
Chapitre 13 - Sénologie -Actualisation 2016 _______________________________________ 104
12.3. A visée thérapeutique _______________________________________________________ 105
12.1.13. Prescription de thérapie ciblée anti HER2 _______________________________________________ 105
12.4. Génotypage utile au diagnostic ________________________________________________ 107
12.5. Génotypage utile au traitement ________________________________________________ 108
Chapitre 14 - Thyroïde– Actualisation 2016 ________________________________________ 109
14.1. Génotypage utile au diagnostic et au traitement __________________________________ 110
14.2. Programme AcSé ____________________________________________________________ 111
14.2.1. Translocation de ALK _______________________________________________________________ 111
14.2.2. Mutation de ALK ___________________________________________________________________ 111
14.2.3. Mutation de MET __________________________________________________________________ 111
Chapitre 15 - Urologie _________________________________________________________ 112
15.1. Cancers du rein– Actualisation 2016 ____________________________________________ 113

15.2. Cancers de la vessie _________________________________________________________ 114
15.3. Carcinomes urothéliaux des voies excrétrices _____________________________________ 114
15.4. Cancers de prostate _________________________________________________________ 114
Chapitre 16 - Annexes _________________________________________________________ 115
16.1. Tableau récapitulatif des examens de biologie moléculaire __________________________ 116
16.2. Charte graphique des arbres décisionnels ________________________________________ 122
16.3. Tables des illustrations _______________________________________________________ 123
16.3.1. Arbres décisionnels _________________________________________________________________ 123
16.3.2. Tableaux _________________________________________________________________________ 123
16.3.3. Figures ___________________________________________________________________________ 124
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
1
/
119
100%