II-4 Les malformations vasculaires

UNIVERSITÉ DE NANTES
FACULTÉ DE PHARMACIE
ANNÉE 2011 N° 2
THÈSE
pour le
DIPLÔME D’ÉTAT
DE DOCTEUR EN PHARMACIE
par
Aurélie Trochu
Présentée et soutenue publiquement le 23 mars 2011
Président :
Mme Céline COUTEAU, Maître de Conférences de Cosmétologie
Membres du jury :
Mme Laurence COIFFARD, Professeur de Cosmétologie
Mme Martine CHAUVELON, Pharmacien
LES « ANGIOMES » ET LEURS TRAITEMENTS,
UNE SOLUTION THÉRAPEUTIQUE D’AVENIR :
LES BÊTABLOQUANTS

Sommaire
Introduction ............................................................................................................................................. 8
Partie I - La peau ................................................................................................................................... 10
I-1 La structure de la peau ................................................................................................................ 11
I-1-1 L’épiderme .......................................................................................................................... 12
I-1-1-1 Les différentes couches de l’épiderme ............................................................ 12
I-1-1-2 Les différents types de cellules et leurs fonctions .......................................... 14
I-1-1-3 La régulation de l’homéostasie épidermique .................................................. 15
I-1-2 Le derme .............................................................................................................................. 15
I-1-2-1 Les couches du derme ..................................................................................... 16
I-1-2-2 Les cellules du derme ...................................................................................... 17
I-1-3 L’hypoderme ....................................................................................................................... 17
I-2 Les annexes cutanées .................................................................................................................. 18
I-2-1 Les poils et les follicules pileux .......................................................................................... 18
I-2-1-1 La structure du poil ......................................................................................... 19
I-2-1-2 La structure du follicule pileux ........................................................................ 19
I-2-2 Les glandes sébacées ........................................................................................................... 21
I-2-3 Les glandes sudoripares ....................................................................................................... 22
I-2-4 Les ongles ............................................................................................................................ 23
I-3 La vascularisation de la peau ....................................................................................................... 24
I-3-1 Le système artériel ............................................................................................................... 25
I-3-2 Le système veineux ............................................................................................................. 26
I-3-3 Les glomus neuro-vasculaires ............................................................................................. 26
I-3-4 Le système lymphatique ...................................................................................................... 26
I-3-5 La formation des vaisseaux sanguins et lymphatiques cutanés ........................................... 26
I-3-5-1 La vasculogenèse ............................................................................................ 27

~ 3 ~
I-3-5-2 L’angiogenèse ................................................................................................. 27
I-3-5-3 La lymphangiogenèse ...................................................................................... 28
I-3-6 La régulation du débit sanguin ............................................................................................ 28
I-4 L’innervation cutanée ................................................................................................................. 29
I-4-1 L’innervation cutanée sensorielle ........................................................................................ 30
I-4-2 L’innervation cutanée végétative ........................................................................................ 31
I-5 Les différentes fonctions de la peau ........................................................................................... 31
I-5-1 La protection de l’organisme ............................................................................................... 31
I-5-2 Le rôle de la peau dans la perception .................................................................................. 33
I-5-3 La thermorégulation ............................................................................................................. 34
I-5-4 Les fonctions métaboliques de la peau ................................................................................ 34
Partie II - Les angiomes ......................................................................................................................... 36
II-1 La classification des angiomes .................................................................................................. 37
II-1-1 Les tumeurs vasculaires ...................................................................................................... 38
II-1-2 Les malformations vasculaires ........................................................................................... 38
II-1-2-1 Les formes isolées à flux lent ........................................................................ 38
II-1-2-2 Les formes isolées à flux rapide .................................................................... 38
II-1-2-3 Les formes complexes ................................................................................... 38
II-2 Les définitions ........................................................................................................................... 39
II-3 Les tumeurs vasculaires .............................................................................................................. 39
II-3-1 Les hémangiomes infantiles ............................................................................................... 39
II-3-1-1 L’épidémiologie ............................................................................................. 39
II-3-1-2 Les aspects cliniques des hémangiomes infantiles ......................................... 40
II-3-1-3 Les diverses localisations des hémangiomes du nourrisson .......................... 41
II-3-1-4 Le diagnostic des hémangiomes infantiles ..................................................... 43
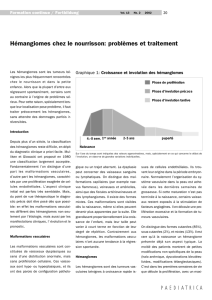
II-3-1-5 L’évolution des hémangiomes infantiles ....................................................... 44

~ 4 ~
II-3-1-6 Les complications des hémangiomes infantiles ............................................. 46
II-3-1-7 La physiopathologie des hémangiomes infantiles ......................................... 48
II-3-2 Les hémangiomes congénitaux .......................................................................................... 49
II-3-3 L’hémangiome en touffe .................................................................................................... 51
II-3-4 L’hémangioendothéliome kaposiforme .............................................................................. 51
II-4 Les malformations vasculaires .................................................................................................. 52
II-4-1 Les malformations capillaires : les angiomes plans ........................................................... 52
II-4-1-1 L’angiome plan : la forme habituelle ............................................................. 52
II-4-1-2 Le syndrome Sturge-Weber-Krabbe .............................................................. 54
II-4-1-3 Le syndrome Klippel-Trenaunay ................................................................... 54
II-4-1-4 Le syndrome de Cobb .................................................................................... 55
II-4-1-5 Les autres malformations capillaires ............................................................. 55
II-4-2 Les malformations veineuses ............................................................................................. 56
II-4-3 Les malformations lymphatiques ....................................................................................... 57
II-4-4 Les malformations artério-veineuses ................................................................................. 58
II-4-5 Les malformations vasculaires complexes ......................................................................... 60
Partie III - Les Traitements ................................................................................................................... 63
III-1 Les traitements des tumeurs vasculaires ................................................................................... 64
III-1-1 Les traitements des hémangiomes infantiles ..................................................................... 64
III-1-1-1 Les traitements classiques pour les formes simples ...................................... 64
III-1-1-2 Les traitements classiques pour les formes graves ........................................ 65
III-1-1-3 Une nouvelle thérapeutique : les β-bloquants ............................................... 73
III-1-2 Les traitements des autres hémangiomes ........................................................................... 80
III-1-2-1 Le traitement des hémangiomes congénitaux R.I.C.H. et N.I.C.H. .............. 80
III-1-2-2 Le traitement de l’hémangiome en touffe .................................................... 81
III-1-2-3 Le traitement de l’hémangioendothéliome kaposiforme .............................. 81

~ 5 ~
III-1-2-4 Le traitement du syndrome de Kasabach-Merritt ......................................... 81
III-2 Les traitements des malformations vasculaires ......................................................................... 82
III-2-1 Les traitements des angiomes plans ................................................................................... 82
III-2-1-1 Le traitement pour la forme habituelle .......................................................... 82
III-2-1-2 Le traitement du syndrome de Sturge-Weber-Krabbe ................................. 84
III-2-1-3 Le traitement du syndrome de Klippel-Trenaunay........................................ 85
III-3 Les traitements des malformations veineuses ...................................................................... 85
III-3-1 La sclérothérapie percutanée ........................................................................... 85
III-3-2 La chirurgie ...................................................................................................... 86
III-3-3 Le laser veineux endovasculaire ...................................................................... 86
III-3-4 Les mesures préventives .................................................................................. 86
III-4 Les traitements des malformations lymphatiques ................................................................ 87
III-4-1 Le traitement de la poussé inflammatoire ........................................................ 87
III-4-2 La chirurgie ...................................................................................................... 87
III-4-3 La sclérothérapie .............................................................................................. 88
III-4-4 Le laser ............................................................................................................ 88
III-5 Les traitements des malformations artério-veineuses .......................................................... 88
Conclusion ............................................................................................................................................. 91
Bibliographie ......................................................................................................................................... 93
Annexes ............................................................................................................................................... 101
Liste des abréviations
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
1
/
117
100%