Synthèse et devenir des ARN

Par Krys3000 (Groupe « The Trust » - http://www.cours-en-ligne.tk/) Page 1
BIOLOGIE MOLECULAIRE
PARTIE II : SYNTHESE ET DEVENIR DES ARN
CHAPITRE I : LA TRANSCRIPTION
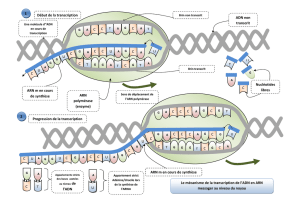
Chez les procaryotes :
La transcription et la traduction s’effectue au même endroit
Lors de la transcription des ARN messagers, on passe directement de l’ADN a l’ARNm
Chez les Eucaryotes :
La transcription (noyau) est séparée de la traduction (cytoplasme)
Il y a des étapes de maturations de l’ARN pré-messager obtenus, aussi appelé ARNnh, en ARNm :
o L’ARNm peut être conçu à partir d’un tas de combinaisons différentes d’exons.
o Il est stabilise par coiffage et ajout d’une queue poly-A
La transcription se fait via une ARN polymérase, de 5’ vers 3’, jusqu’a un terminateur qui la fait se détacher (voir génie
génétique).
I MECANISTIQUE DE TRANSCRIPTION
L’ARN polymérase est compose de plusieurs sous-unités qui chez les procaryotes ont chacune des caractéristiques et rôles
différents :
• α se lie aux séquences de régulation (il y en a deux)
• β et β’ forment des ponts phosphodiesters
• ω permet d’assembler les sous-unités (notamment β et β’)
Ensemble, cela forme l’Enzyme Core, qui doit se lier a une dernière sous-unité pour former une holoenzyme spécifique : σ. C’est
cette sous-unité qui donne la spécificité a l’enzyme pour le promoteur et lui permet de démarrer pour rechercher des séquences
spécifiques : la région « -35 » (appelée ainsi car située généralement 35 pb avant le début de la transcription), suivi de la
Pribnow Box ou séquence « -10 », 25 paires de bases plus loin. Elle va alors se fixer, puis σ se détache et la transcription peut
commencer a partir d’un point situe 10 pb plus tard : le point « +1 ».
On passe ensuite a la phase d’élongation de la transcription, ou la « bulle » de transcription de 17 pb, formée par l’enzyme et
l’ADN, se déplace, déroulant l’ADN pour transcrire une séquence d’ARN. Cette opération entraine, par l’action de
topoisomérases, une compensation par un super enroulement de l’ADN, positif en aval, et négatif en amont, de la séquence
transcrite.
Enfin, vient la phase de terminaison. Il y a deux types de terminaison. Dans les deux cas, le terminateur est une structure qui se
met en boucle lorsqu’elle est au contact de la polymérase (voir génie génétique) :
• Terminaison « Rho-indépendante » - exposée à la boucle, la polymérase marque un temps de pause. Déstabilisée, elle
lâche sa prise et quitte la séquence d’ADN.
• Terminaison « Rho-dépendante » - exposée a la boucle, la polymérase marque un temps de pause, mais tiens bon. Ce
battement suffit cependant a la protéine Rho, hexamère qui se lie des le départ a l’ARN en cours de formation sur une
séquence riche en C et pauvre en G, et qui ≪ poursuit ≫ la polymérase, pour rattraper celle-ci, et forcer le décrochage.
Le contrôle de l’expression génique est sous la direction des régions de contrôle génique, terme utilise pour décrire les
séquences régulatrices ou « éléments cis-régulateurs » et le promoteur du gène. Il s’effectue par la présence de protéines
régulatrices ou « éléments trans-régulateurs » et de facteurs de transcription généraux. Ces séquences peuvent appartenir a
deux catégories : les enhancers et les silencers. Ce sont des séquences d’ADN qui peuvent respectivement activer ou réprimer
l’activité du promoteur. Elles peuvent être situées n’ importe où (5’UTR, ORF, 3’UTR, même parfois très loin du promoteur !) et
régulent l’expression du gène spécifiquement dans un tissu ou au cours du développement, dans les deux sens. Elles sont

Par Krys3000 (Groupe « The Trust » - http://www.cours-en-ligne.tk/) Page 2
également capables de moduler l’activité temporelle et spatiale de n’importe quel gène si elles sont fusionnées en amont ou en
aval du gène.
II LES DIFFERENTES POLYMERASES
Chez les procaryotes il n’existe qu’un seul type d’ARN Polymérases, mais ce n’est pas le cas chez les Eucaryotes qui en disposent
de 3 types, qui ne synthétisent pas les mêmes types d’ARN :
Polymérases I (nucléole – insensible aux toxines) : Précurseurs d’ARNr et ARNr cytoplasmiques
Polymérases II (nucleoplasme – sensibles aux fortes concentrations de toxines) : ARNnh, ARNm
Polymérases III (nucleoplasme – très sensible aux toxines) : petits ARN (ARNt)
A] POLYMERASE I
Les promoteurs qui vont être reconnus par les polymérases de types I ont deux composants de séquences : le « core » du
promoteur qui comprends les séquences régulatrices évoquées plus haut (de -45 a +20 environ) et les éléments de contrôles
(UCE) situes environ de -180 a -107. Ces éléments ne sont pas nécessaires mais augmentent l’activité.
Deux facteurs entrent en jeu :
UBF1 qui se fixe sur des séquences GC a la fois dans l’UCE et sur le core
SL1 se fixe à UBF1 a la fois dans l’UCE et sur le core
La polymérase peut alors se positionner sur le core et démarrer. On ignore encore comment et pourquoi le fait d’avoir
cette configuration sur l’UCE également augmente l’activité de la polymérase.
Les ARN ribosomaux obtenus vont alors subir un épissage afin d’obtenir les différents types d’ARNr qui composeront le
ribosome.
B] POLYMERASE III
Contrairement a la polymérase de type I, il existe 3 types de promoteurs pour cette polymérase :
• Le promoteur de type 1, compose de deux séquences en aval du point +1 : la Box A et la Box C.
• Le promoteur de type 2, compose de deux séquences en aval du point +1 : la Box A, immédiatement âpres le point +1,
et la Box B, quelques bases plus loin.
• Le promoteur de type 3, compose de trois séquences en amont du point +1 : Une TATA Box (qui va jouer exactement le
même rôle que la Pribnow Box des Procaryotes) juste avant le promoteur, laquelle suffit à l’expression du gène, mais
également les séquences PSE, située un peu avant, et Oct qui se trouve tres loin en arrière du +1. Ces deux dernières
séquences augmentent l’activité du promoteur.
De la même manière que chez les polymérases de type I, toutes ces séquences servent à lier des facteurs. Il existe donc les
facteurs généraux, nécessaires au mécanisme en lui-même, qui peuvent se fixer en amont comme en aval du site d’initiation
(+1), les facteurs ubiquitaires se fixant en amont, mais proche du site d’initiation, augmentant l’efficacité, et les facteurs
inductibles, également en amont, mais loin du site d’initiation, régulant l’expression des gènes.
C] POLYMERASE II
La polymérase de type II est responsable de la synthèse des ARNnh (et donc des ARNm). Afin de déterminer la localisation du
promoteur, on peut employer la technique du run-off assay, une technique de transcription in vitro :
1. On digère un fragment linéaire de l’ADN, lequel possède un promoteur de polymérase type II dont on veut connaitre la
position, avec une enzyme de restriction : le site correspondant doit se trouver dans un emplacement connu de la
région codante du gène.
2. On met au contact notre gène tronqué avec une ARN polymérase II, des facteurs « ad hoc » ou un extrait partiellement
purifié, et des rXTP marqués.
3. La polymérase avance et arrive à l’ endroit de la coupure ou elle se retrouve donc éjectée dans le vide, formant des
fragments incomplets.
4. On récupère le fragment d’ARN marque. Sachant l’emplacement du site dans la région codante, il suffit de regarder la
taille du transcrit obtenu pour retrouver l’emplacement du point +1.
Cette technique peut être utilisée pour tester l’importance des protéines.

Par Krys3000 (Groupe « The Trust » - http://www.cours-en-ligne.tk/) Page 3
La plupart des promoteurs PolII possèdent une TATA Box (-25 a -35) et un élément Inr (a cheval sur le +1). Certains ne possèdent
qu’un seul des deux et d’autres encore carrément aucun des deux. Il existe également une autre séquence, celle-ci se trouvant
en aval du site d’initiation cette fois : c’est la séquence DPE pour Downstream Promotor Element.
III LES FACTEURS GENERAUX DE LA TRANSCRIPTION
Comme évoqué précédemment, la synthèse d’ARN, toutes polymérases confondues, se fait en présence de facteurs qui se lient
sur les différences séquences (ou sur d’autres facteurs). Parmi eux, les facteurs généraux, qui sont obligatoire pour avoir
transcription. Ceux-ci vont en effet former, avec la Polymérase elle-même, le Complexe d’initiation de la Transcription.
Dans le cas des ARN polymérases de types I et III, il n’existe que peu de facteurs généraux. En revanche, la polymérase de type II,
celle qui nous intéresse le plus car à l’origine des ARNm et donc des protéines, dispose d’une multitude de facteurs :
Le premier à entrer en action est le TFIID, un complexe qui réunit en fait deux facteurs : une sous-unité de TBP qui va venir se
fixer a la TATA Box, et 12 sous-unités de TAF, dont le rôle est d’interagir avec l’Inr et le DPE afin de « brancher » TBP sur la TATA
Box. Toutefois, cette fixation ne peut se faire qu’a l’aide d’un autre facteur : TFIIA, facteur en 3 sous-unités qui dissocie le dimère
de TBP, dont la forme dimérique « de base » n’est pas compatible à un branchement sur l’ADN.
Le facteur TFIID dispose d’un second rôle – celui de recruter TFIIB (1 sous-unité), facteur qui va sélectionner le site de démarrage
de la polymérase, et recruter celle-ci en plus d’un 4
ème
facteur, TFIIF (2 sous-unités). Le rôle de TFIIF va être majoritairement de
cibler la polymérase sur le promoteur et de détruire toutes les interactions non spécifiques « parasites » qui pourraient se
former. A son tour, il appelle alors le facteur suivant, TFIIE (2 sous-unités). Celui-ci n’a pas d’autre rôle que de recruter TFIIH et
moduler les activités de celui-ci, des activités kinase, hélicase et ATPase, qui vont permettre la libération du promoteur (par
phosphorylation du domaine C-Terminal) et l’avancée de la polymérase.
IV LES TECHNIQUES D’ANALYSE
Il existe plusieurs techniques que l’on utilise pour étudier les acides nucléiques.
Technique de Southern Blot (permet d’identifier de l’ADN de le séparer selon son poids moléculaire)
Migration sur gel en conditions non-dénaturantes d’ADN colore au BET, par le biais d’une électrophorèse.
Le gel est place sur une éponge, laquelle baigne dans une solution saline. On pose une membrane de nitrocellulose sur
le gel, et du papier absorbant par-dessus.
Récupération de la membrane sur laquelle l’ADN a filtré et a été transféré. Hybridation dans un sac spécifique avec une
sonde.
Lavage de la membrane pour éliminer l’excès de sonde.
Autoradiographie et obtention d’un autoradiogramme sur lequel la sonde s’est liée de façon complémentaire aux ADN
étudiés.
Technique de northern Blot (permet d’identifier de l’ARN de le séparer selon son poids moléculaire)
Migration sur gel contenant du formaldéhyde comme dénaturant d’ARN colore au BET, par le biais d’une
électrophorèse.
Le gel est place sur une éponge, laquelle baigne dans une solution saline. On pose une membrane de nitrocellulose sur
le gel, et du papier absorbant par-dessus.
Récupération de la membrane sur laquelle l’ARN a filtré et a été transféré. Hybridation dans un sac spécifique avec une
sonde.
Lavage de la membrane pour éliminer l’excès de sonde.
Autoradiographie et obtention d’un autoradiogramme sur lequel la sonde s’est liée de façon complémentaire aux ARN
étudiés.
Technique de la RT-PCR (permet d’amplifier un fragment d’ARN sous forme d’ADNc en combinant une transcription inverse et
une PCR)
Etape de transcription inverse : utilisation d’un reverse transcriptase pour passer de l’ARNm a un hybride ARNm/ADNc,
puis d’une RNAse H pour couper le brin d’ARN, et d’une ADN Polymerase pour détruire ce brin et synthétiser un brin
d’ADNc (voir le cours de génie génétique).
PCR : Amplification du brin d’ADNc (plus stable que l’ARN) avec des amorces, une Taq polymérase (résistante a la
chaleur) et des dNTP, selon les 3 étapes classiques d’une PCR sur n cycles – dénaturation des brins par portage a 94°C,
hybridation des amorces aux brins isoles (possible uniquement si la température est baissée a 55°C), puis
polymérisation (a 72°C, température optimale de la polymérase).

Par Krys3000 (Groupe « The Trust » - http://www.cours-en-ligne.tk/) Page 4
On obtient alors, après n cycles de PCR, 2
n
fois la quantité d’ADNc de départ. Cette version d’analyse de PCR est dite « PCR en
point final » du fait que l’on obtient les données à la fin des n cycles.
Technique de qPCR (permet d’évaluer en temps réel la quantité d’ADN forme par PCR)
Cette technique, aussi appelée « PCR en temps réel » permet de détecter et
quantifier l’amplification de l’ADN à l’aide d’un marqueur fluorescent et du
suivi de la quantité de fluorescence a chaque cycle. On utilise comme
marqueur, par exemple, le SYBR Green, un agent qui se lie a l’ADN double
brin et devient plus fluorescent lorsqu’il est lié. On obtient ce genre de
courbe, et l’on peut ainsi déterminer la valeur du cycle seuil, Ct. Celle-ci
dépend du nombre de matrices au départ : plus il y a de matrices à
amplifier, plus on arrivera vite au seuil Ct.
Cela nous permettra de quantifier relativement les ARNm sans courbe de
standard, en comparant simplement les Ct, avec l’aide d’un gène
domestique qui nous servira de contrôle endogène (vérificatif du dosage de
l’ARN, de la qualité de celui-ci, et de l’efficacité de la Reverse Transcription) et d’un calibrateur (qui représente l’état normal
physiologiquement – c’est le standard).
Cartographie a la RNAse (permet d’analyser un fragment d’ARN)
Cela nécessite d’avoir déjà un morceau de l’ADN correspondant.
Insertion dans un vecteur que l’on va ensuite faire exprimer avec une RNA Polymérase sp6 et des rNTP marqués, pour
obtenir de l’ARN marqué qui nous servira de ribosonde.
Hybridations de nos fragments d’ARN avec les ribosondes obtenues
Digestion par RNAse A, enzyme qui dégrade spécifiquement l’ARN simple brin, pour conserver uniquement le fragment
accroché à la sonde.
Analyse sur gel en condition dénaturante suivie d’une autoradiographie. Le fragment digéré par RNAse va plus loin que
le fragment non digéré.
Transcription sur noyaux isoles (Run-on) (permet de déterminer le type de régulation d’un gène étudié)
Lyse ménagée de cellules afin de récupérer les noyaux isolés.
Incubation des noyaux 5 minutes a 30 °C avec des XTP marques.
Ajout d’héparine, qui stoppe l’initiation de transcription. Subsiste alors les ARN dont la transcription a débuté in vivo
avant la lyse. Ceux-ci vont donc continuer leur élongation de façon in vitro.
Purification et récupération des ARN marqués qui vont servir de sonde
Hybridation avec les ADN correspondants aux gènes étudiés fixes sur membrane. Selon le résultat du run-on et d’un
northern complémentaire, on peut alors déterminer le type de régulation :
o Si le run-on ne nous montre aucune décroissance de la transcription, tout comme le northern, c’est qu’il n’y a
pas de régulation.
o Si le run-on ne nous montre aucune décroissance de la transcription, mais que le northern montre qu’il y a de
moins en moins d’expression, c’est que la régulation du gène est post-transcriptionnelle.
o Si le run-on nous présente une décroissance au cours du temps, et que le northern nous montre que le gene
est de moins en moins exprime, c’est que la régulation est transcriptionnelle.
o Si le run-on nous présente une décroissance au cours du temps, et que le northern nous montre un arrêt de
l’expression brutal et rapide, c’est que la régulation est à la fois transcriptionnelle et post-transcriptionnelle.
Digestion a la nucléase S1 (permet de trouver le 5’ d’un transcrit)
Marquage d’un brin d’ADN en 5’.
Hybridation du brin avec l’ARN correspondant.
Digestion du surplus avec une nucléase S1, enzyme digérant tout ce qui est simple brin (ADN et ARN).
Dénaturation et autoradiographie du brin d’ADN.
Extension d’amorce (permet de trouver le 5’ d’un transcrit)
Marquage d’un oligonucléotide en 5’.
Hybridation de l’oligonucléotide avec notre ARN.
Reverse Transcription (avec Reverse Transcriptase + dNTPs).
Dénaturation et autoradiographie du brin d’ADN.
RACE PCR (permet de trouver le 5’ ou le 3’ d’un transcrit)
A l’aide d’une ligase, lier un adaptateur en 5’ sur l’ARNm.

Par Krys3000 (Groupe « The Trust » - http://www.cours-en-ligne.tk/) Page 5
Réaliser une Reverse Transcription pour obtenir un hybride ARNm/ADNc
Digérer l’ARN a la RNAse.
Lier un autre adaptateur en 5’ de l’ADNc.
1
er
tour de PCR avec un OligodT en 5’.
2
ème
tour de PCR avec un OligodT spécifique en 5’ sur l’autre brin
N tours de PCR, puis recommencer avec deux autres oligos (un sur chaque brin) décalés par rapport aux premiers.
Technique du gène rapporteur (permet de déterminer les séquences régulatrices importantes en 5’UTR d’un promoteur)
Introduction d’un « gène rapporteur » dans la séquence génétique, en aval de la région 5’ étudiée, via transfection,
introduction par vecteur viral ou transgénèse. Selon le type de détection, on emploiera :
o Enzymatique : β-galactosidase, luciferase, CAT
o Immunologique : TSV40, β-globine
o Fluorescente : GFP
Mesure de l’activité de transcription du gène rapporteur lorsque l’on supprime différentes parties du 5’UTR étudié. Les
parties qui, lorsqu’elles sont supprimées, détruisent ou faiblissent sévèrement l’activité transcriptionnelle sont les
parties essentielles.
Protection a la DNAse I (footprint) (permet de localiser et identifier les sites de liaisons Protéines/ADN)
Marquage de fragment d’ADN (amplifié par PCR par exemple)
Constitution de deux pools : un avec de l’ADN marque, l’autre avec de l’ADN marque mis au contact de protéines de
liaisons
Digestion partielle à la DNAse I des deux pools
Electrophorèse/Autoradiographie. Les fragments manquants à l’image sur le pool mis au contact avec les protéines sont
les résultantes du fait que les protéines étaient liées à ces endroits la, protégeant ainsi l’ADN de la digestion. Ce qui
permet de connaitre l’emplacement de la séquence de liaison.
Méthode de retard sur gel (EMSA) (permet de vérifier l’interaction entre une protéine et l’ADN)
Marquage d’ADN
 6
7
8
9
6
7
8
9
1
/
9
100%