Cours de pharmacologie

Université D’El-Oued Année 2014/2015
Cours de pharmacologie 3ème Année Biochimie, licence LMD
1|Page
Enseignant M. MEDJOUR A.
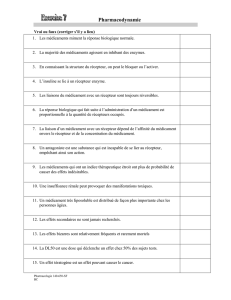
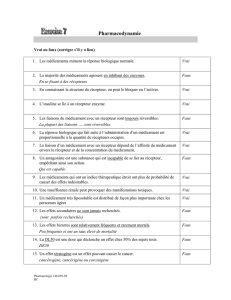
1. Généralités
1.1. Introduction
L’objet de la pharmacologie est d’étudier le devenir de médicament dans l’organisme
depuis sa prise par le malade jusqu’à son effet thérapeutique puis son élimination.
Le terme vient de Grec pharmakon, « médicament », et logos, « disscours ». Alors la
pharmacologie est la science des médicaments.
1.2. Domaine et limites de la pharmacologie
La pharmacologie est une discipline carrefour qui touche à la pharmacie, la chimie, la biologie,
la génétique, la pathologie, la thérapeutique et à bien d’autres sciences. Elle-même se subdivise
en spécialités multiples :
pharmacologie moléculaire ;
pharmacocinétique : devenir des médicaments au sein des organismes
vivants ;
pharmacodynamie : effets des médicaments sur les systèmes
biologiques ;
dosage des médicaments et suivi thérapeutique ;
usage des médicaments en médecine humaine ;
chronopharmacologie : médicaments et cycles biologiques ;
pharmacologie clinique : médicaments et êtres humains ;
essais thérapeutiques : expérimentation des médicaments chez l’homme ;
pharmacovigilance : effets indésirables des médicaments ;
intoxications médicamenteuses : effets des surdosages ;
pharmaco-épidémiologie : médicaments et populations ;
pharmaco-économie : économie du médicament ;
pharmacogénétique : génome et médicament ;
pharmacologie sociale : société et médicament ;
sans compter les pharmacologies spécialisées aux classes pharmaco-
thérapeutiques de médicaments et etc.
1.3. Définition, origine et nomenclature des médicaments
1.3.1. Définition
Règlementaire
On entend par médicament toute substance ou composition présentée comme possédant
des propriétés curatives ou préventives, à l’égard des maladies humaines ou animales, ainsi que

Université D’El-Oued Année 2014/2015
Cours de pharmacologie 3ème Année Biochimie, licence LMD
2|Page
Enseignant M. MEDJOUR A.
tout produit pouvant être administré à l'homme ou à l'animal en vue d'établir un diagnostic
médical ou de restaurer, corriger ou modifier leurs fonctions organiques.
Pharmacologique
Un médicament est constitué d’un principe actif et de « substances inertes » adaptées à
la voie d’administration à laquelle il est destiné et que l'on appelle excipient. La substance active
est présente à une concentration appelée « dosage ». La substance active possède des propriétés
pharmacologiques qu'elle exerce sur une cible organique ou fonctionnelle, d'où résulteront les
indications thérapeutiques.
1.3.2. Origine
Les trois règnes de la nature (végétal, animal et minéral) et aussi la voie synthétique
fournissent des principes actifs pouvant conduire à des médicaments.
1.3.2.1. Origine végétal
C’est la source la plus ancienne, mais qui reste d’actualité (on recherche toujours des
«principes actifs» dans les recettes de « médecine traditionnelle » ou de façon systématique
dans les extraits végétaux).
Il est classique de distinguer parmi les produits végétaux :
Les alcaloïdes (littéralement, « comme les alcalins ») ex : quinine, strychnine, émétine,
morphine, papavérines, réserpines, etc. ;
Les gommes ; ex. : mucilages laxatifs, gommes pour suspension (arabique, adragante) ;
Les glycosides (ils contiennent des sucres dans leurs structures chimiques) ; ex. :
digitoxine, digoxine.
1.3.2.2. Origine animale
Extrait du sang humain. Ex. : fibrinogène, prothrombine, proconvertine, facteurs
Stuart et anti-hémophilique B (PPSB).
Hormones polypeptidiques extractives. Ex. : insulines, gonadotrophines.
Enzymes. Ex. : trypsine, chymotrypsine, Kinases (urokinase, streptokinase).
Substances diverses obtenues par techniques de « génie génétique ». Ex. :
interféron, interleukines, insulines, hormones de croissances, etc.
Excipients pharmaceutiques. Ex. : lanoline, axonge.
1.3.2.3. Origine Synthétique
La plupart des médicaments actuellement commercialises sont d’origine synthétique. Ils
sont obtenus par :
- Synthèse totale ou

Université D’El-Oued Année 2014/2015
Cours de pharmacologie 3ème Année Biochimie, licence LMD
3|Page
Enseignant M. MEDJOUR A.
- hémi-synthèse ; ex. : certaines pénicillines : dans ce cas, une chaine latérale est greffée sur
une structure de base fournie par un organisme vivant et cette adjonction confère à la molécule
des propriétés nouvelles, comme une résistance aux pénicillinases.
1.3.2.4. Origines biogénétiques
Les méthodes de génie génétique sont les dernières venues parmi les méthodes
d’obtention des médicaments : elles permettent de faire fabriquer par des cellules vivantes
(procaryotes ou eucaryotes) des substances naturelles polypeptidiques présentant toutes les
caractéristiques de leur modèle humain, puisque le code génétique a été établi, puis incorpore
dans le génome des cellules sources ; celles-ci, mises en culture, se multiplient ensuite à l’infini
en produisant la substances dont elles portent le code.
La production de masse de ces protéines parfaitement définies a permis d’obtenir de
nouveaux médicaments : des hormones (hormones de croissance [GH], insulines dite
« humanisée » ou modifiées [« lispro », pour lysine-proline]), des facteurs de croissance
hématopoïétiques (érythropoïétine [EPO], G-CSF, GM-CSF, etc.) des cytokines
(interleukine-2 [IL-2], les différents types d’interférons) des médicaments procoagulants (tel le
facteur VIII ou facteur anti-hémophilique A). Citons aussi les anticorps monoclonaux.
1.3.3. Nomenclature (Dénomination) des médicaments
Chaque médicament possède au moins trois noms.
A. Nom chimique
Qui est élaborée à l’aide des règles de nomenclatures très strictes contrôlées par un
organisme l’IUPAC (= International Union of Pure and Applied Chemistry).
Exemple :
Phényléthylmalonylurée ;
Dicarbamate de méthylpropylpropanediol.
B. Dénomination commune internationale «DCI»
Exemple (pour les noms chimiques cites en exemples plus haut)
- Phénobarbital ;
- Méprobamate.
Cette DCI est attribuée par l’OMS (organisation mondiale de la santé) qui est un
organisme international indépendant des firmes pharmaceutiques, selon des directives
générales permettant d’exclure toute influence commerciale pour le choix du nom, et permettant
de regrouper selon des assonances voisines, des produits appartenant à la même classe
pharmacologique.

Université D’El-Oued Année 2014/2015
Cours de pharmacologie 3ème Année Biochimie, licence LMD
4|Page
Enseignant M. MEDJOUR A.
Exemple la DCI des inhibiteurs de l’enzyme de conversion : captopril, enalapril, lisinopril,
perindopril, quinapril, ramipril, benazeprilet etc
C. Nom de spécialité ou nom de marque
Exemple (toujours pour les deux mêmes produits) :
Phénobarbital ou GARDENAL ; Méprobamate ou EQUANIL.
(Le signe * ou le signe « R » veut dire « registré = marque déposée », car ce nom est une
propriété commerciale).
Dans ce domaine du nom de spécialité, l’imagination est reine et la création d’un nom de
marque se réfère aux seuls impératifs commerciaux : il s’agit de faire prescrire le médicament,
de préférence à son concurrent, à la fois en frappant de façon consciente ou inconsciente l’esprit
du médecin par une représentation symbolique et flatteuse de son efficacité et de sa sécurité
d’emploi, et en aidant la mémoire du prescripteur ;
1.4. Les différents types de médicaments
L’étude de la préparation des médicaments s’appelle pharmacie galénique (un mot tiré de
Galien, médecin grec du IIe siècle). On distingue :
Les spécialités pharmaceutiques (environ 9 000 en France) sont préparées par
l’industrie pharmaceutique. Elles doivent obtenir une autorisation de mise sur le marché
(AMM).
Les génériques. Après 20 ans de commercialisation, un médicament tombe dans le
domaine public, c’est-à-dire que tout laboratoire peut le fabriquer et le commercialiser.
Le nom du médicament est alors le nom de substance active (et pas celui d’une
spécialité). Le générique est une copie conforme de la spécialité originale, mais à un
prix inférieur.
Les préparations magistrales très rares, sont réalisées par le pharmacien à partir d’une
formule rédigée par un médecin pour un seul malade.
Les médicaments officinaux sont préparés par le pharmacien à partir d’une formule
inscrite sur un formulaire officiel.
2. Pharmacocinétique
La pharmacocinétique : a pour but d’étudier le devenir du médicament dans l’organisme.
On peut schématiser la pharmacocinétique d’un médicament en 4 grandes étapes (résorption
ou absorption, distribution, métabolisme, élimination de l’organisme) « RDME ».
2.1. Résorption
La résorption est le processus par lequel le médicament passe dans la circulation
générale depuis son site d’administration.

Université D’El-Oued Année 2014/2015
Cours de pharmacologie 3
ème
Année Biochimie, licence LMD
5|Page
Enseignant M. MEDJOUR A.
L’étape de la résorption n’existe pas lorsque le principe actif (PA) est administré par
voie intraveineuse.
Les paramètres pharmacocinétiques qui permettent de quantifier le processus de résorption
sont :
- le coefficient de résorption, définit comme la fraction du médicament administré qui franchit
la membrane gastro-intestinale.
- La biodisponibilité se définit par la quantité de principe actif qui parvient à son site d’action
et la vitesse avec laquelle il y accède.
La mesure au niveau du site d’action étant difficile à obtenir, on considère plus
généralement la biodisponibilité comme la fraction du médicament qui atteint la circulation
générale.
2.1.1. Facteurs de variation de la résorption digestive
a) forme galénique : La galénique, c’est à dire la forme sous laquelle se trouve le médicament
(le principe actif + des excipients), joue un rôle important dans les différentes phases qui
conduisent à la solubilisation du médicament, condition indispensable à sa résorption.
Il existe des formes galéniques particulières dont la dissolution répond à des cinétiques
spécifiques :
- forme à libération prolongée (LP) : libère une quantité constante de médicament par unité de
temps. Ceci permet de maintenir plus longtemps les concentrations plasmatiques dans la zone
d’efficacité thérapeutique.
Exemple : Alpress LP® est une formulation qui permet de maintenir les concentrations
plasmatiques en plateau de la 6e à la 24e heure après l’administration d’un comprimé.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
1
/
26
100%