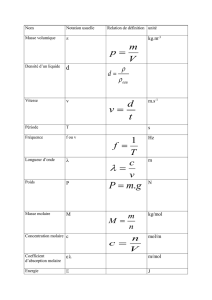
4Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
Programme
I - EXPLORATION DE L’ESPACE (5 TP, 10 heures en classe entière)
EXEMPLES D’ACTIVITÉS CONTENUS COMPÉTENCES EXIGIBLES
Expériences mettant en évidence qualitativement
des transformations lentes et rapides et les facteurs
cinétiques, température et concentration à l’aide
d’observations visuelles : H2O2 + I– et S2O3
2– + H+,
tests caractéristiques utilisant le réactif de Fehling,
le réactif de Tollens, par exemple.
– d’un capteur de pression, d’une balance, d’un
conductimètre, etc.
Illustrations dans la vie courante : cuisson à l’auto-
cuiseur, conservation des aliments par le froid, etc.
1. Transformations lentes et rapi-
des
– Mise en évidence expérimentale
de transformations lentes et rapides.
– Mise en évidence expérimentale
des facteurs cinétiques : tempéra-
ture et concentration des réactifs.
– Rappels sur les couples oxydant/
réducteur et sur l’écriture des équa-
tions de réactions d’oxydoréduction.
– Écrire l’équation de la réac-
tion associée à une transformation
d’oxydoréduction et identifier dans
cette équation les deux couples mis
en jeu.
– Définir un oxydant et un réduc-
teur.
– Montrer, à partir de résultats expé-
rimentaux, l’influence des facteurs
cinétiques sur la vitesse de réaction.
Transformations lentes et rapides
1
PARTIE 1
LA TRANSFORMATION D’UN
SYSTÈME CHIMIQUE EST-
ELLE TOUJOURS RAPIDE ?
Cours
Découpage du cours
1. Rappels d’oxydoréduction p. 14
2. Cinétique chimique p. 15
3. Transformations rapides p. 16
4. Transformations lentes p. 18
5. Facteurs cinétiques p. 19
6. Applications p. 20
Ce chapitre présente tout d’abord des rappels d’oxy-
doréduction de la classe de 1re S, car la partie du
programme « La transformation d’un système
chimique est-elle toujours rapide ? » s’appuie essen-
tiellement sur des transformations chimiques met-
tant en jeu l’oxydoréduction.
♦Les élèves n’ont jusqu’alors rencontré que des exemples
de transformations rapides, par une approche simple
s’appuyant largement sur des observations expérimen-
tales : ils vont appréhender des systèmes chimiques
dont les évolutions temporelles sont différentes.
1. Rappels d’oxydoréduction
On évoque l’oxydoréduction par le biais de rappels
de définitions : oxydants, réducteurs, couples oxy-
dant/réducteur, réaction d’oxydoréduction.
2. Cinétique chimique
L’évolution temporelle des systèmes chimiques
n’ayant jamais été traitée dans les classes précé-

5
Chapitre 1 - Transformations lentes et rapides
décomposition d’une solution aqueuse de perman-
ganate de potassium. Cette lente réduction des ions
permanganate par l’eau est accélérée en milieu
acide. Pour conserver le titre d’une solution aqueuse
de permanganate de potassium, il est impératif de la
préparer au dernier moment (solution fraîche) et
surtout de ne pas l’acidifier lors de cette préparation
(l’acidification avec de l’acide sulfurique s’effectue
dans le vase réactionnel).
5. Facteurs cinétiques
Les grandeurs modifiant la durée d’évolution d’un
système chimique sont abordées qualitativement
dans cette fin de chapitre.
Conformément au programme, seules la concentra-
tion des réactifs et la température sont abordées.
Mais dans l’activité 4, l’occasion est donnée d’évo-
quer l’état de division des solides, un facteur cinéti-
que largement mis à profit dans les expériences avec
les métaux au collège.
6. Applications
Il s’agit d’illustrations prises dans la vie courante :
diminution d’un temps de cuisson avec un autocui-
seur, conservation des aliments par baisse de tem-
pérature.
Dans la classe, on évoque la diminution de la durée
d’une transformation chimique avec un montage à
reflux et le blocage d’une telle transformation par
trempe chimique ou physique.
dentes (toutes les transformations étudiées étant
quasi immédiates), le programme de TS aborde un
nouveau domaine de la chimie.
Pour la distinction entre transformations lentes et
rapides, la persistance rétinienne constitue un réfé-
rent temporel.
3. Transformations rapides
Ces transformations chimiques, mettant en jeu des
réactions de combustion, acido-basiques, de préci-
pitation et surtout des réactions d’oxydoréduction
très visuelles (iodométrie, manganimétrie), per-
mettent de reprendre les bases de deux chapitres du
cours de 1re S.
4. Transformations lentes
La formation de rouille, étudiée en classe de troi-
sième, constitue une bonne introduction aux trans-
formations lentes. Les réactions d’oxydoréduction
sont en effet des supports très visuels pour les trans-
formations chimiques à évolution lente. Ainsi l’ac-
tion de l’eau oxygénée sur les ions iodure se traduit
par l’apparition progressive de la couleur brune du
diiode qui se forme.
Le test au miroir d’argent s’appuie sur une transfor-
mation chimique lente au cours de laquelle il y a
réduction des ions argent (I) complexés en milieu
basique.
Transformation extrêmement lente, l’action des
ions permanganate sur l’eau conduit lentement à la
Activités
2. L’état final dans les deux béchers se caractérise
par une décoloration, la réduction des ions per-
manganate conduit en milieu acide aux ions man-
ganèse (II) incolores, les autres espèces étant
également incolores.
Généralement, la transformation dans le bécher 2
est terminée au bout de 5 min.
3. La transformation dans le bécher 1 est quasi ins-
tantanée ; elle est lente dans le bécher 2.
4. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l)
Fe
2+(aq) = Fe3+(aq) + e–.
Les deux demi-équations mettent en évidence un
transfert d’électrons.
5. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l)
C2O4H2(aq) = 2 CO2(aq) + 2 H+(aq) + 2 e–.
On retrouve ainsi l’équation de la réaction en mul-
tipliant la première demi-équation par 2 et la
seconde par 5 :
Transformations rapides
ou lentes ? (page 22)
Cette activité permet d’aborder la cinétique des
transformations à travers l’iodométrie et la manga-
nimétrie, puis d’écrire des demi-équations des cou-
ples utilisés. On présente également des espèces
colorées (ion permanganate et diiode) qui permet-
tent de bien appréhender visuellement les évolutions
lentes ou rapides des systèmes chimiques étudiés. Il
est intéressant d’observer qu’un même couple
oxydo-réducteur peut, selon le partenaire, partici-
per à des transformations lentes ou rapides.
➜ A Manganimétrie
1. La transformation immédiate se traduit par une
décoloration dans le bécher 1.
Dans le bécher 2, la réduction des ions permanga-
nate n’est pas immédiate, la couleur rose peut per-
sister plusieurs minutes.
A
C
T
I
V
I
T
É
1
A
C
T
I
V
I
T
É
1

6Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
2 MnO4
–(aq) + 5 C2O4H2(aq) + 6 H+(aq)
2 Mn2+(aq) + 10 CO2(aq) + 8 H2O(l).
➜ B Iodométrie
1. a) 2
S2O3
2–(aq)
=
S4O6
2–(aq)
+ 2 e–
I
2(aq) + 2 e– = 2 l–(aq)
2
S2O3
2–(aq)
+ I2(aq) S4O6
2–(aq)
+ 2 l–(aq).
b) Oxydation
I2(aq) + 2 S2O3
2–(aq)
2 l–(aq) + S4O6
2–(aq).
Réduction
2. Il y a réduction du diiode (espèce colorée) par les
ions thiosulfate avec formation de deux ions inco-
lores. La transformation chimique se caractérise
par une décoloration quasi immédiate.
3. Les réactifs sont des espèces chimiques incolo-
res : il se forme du diiode (espèce colorée) et des
ions sulfate (incolores). La formation de diode per-
met donc le suivi temporel de la transformation.
4. La formation progressive de diiode se traduit par
une intensification de la couleur jaune dans le
bécher, dont la solution présente une teinte brune
en fin de transformation.
Mise en évidence
de facteurs cinétiques (page 23)
À l’aide d’expériences d’oxydoréduction, cette acti-
vité permet de mettre en évidence, par une observa-
tion qualitative et probante, la notion de facteur
cinétique.
➜ A Action de l’eau oxygénée H2O2(aq)
sur les ions iodure I–(aq)
1. Le diiode (espèce chimique colorée) formé lors
de la transformation chimique permet de suivre
temporellement la transformation.
2. a) Les six tubes côte à côte simulent des avance-
ments différents de la réaction. Le contenu du tube
1 est plus foncé que celui des tubes 2, 3, 4, 5 et 6.
Celui du tube 2 plus foncé que le 3, lui-même plus
foncé que le 4, etc.
b) Le suivi visuel du contenu des six tubes montre
que la transformation est lente et que la formation
progressive de diiode intensifie peu à peu la cou-
leur du contenu du tube.
3. Les systèmes chimiques sont initialement iden-
tiques ; l’état final de chaque système conduit à la
formation de la même quantité de diiode. Les
A
C
T
I
V
I
T
É
2
A
C
T
I
V
I
T
É
2
contenus des six tubes sont identiques en fin de
transformation.
➜ B Modifier la durée d’évolution
1. a) Le contenu de l’erlenmeyer (2) présente une
teinte plus foncée que celui de l’erlenmeyer (1).
b) Le système chimique contenu dans l’erlenmeyer
(2) évolue plus rapidement que le (1). La couleur de
la solution est liée à la formation progressive de
diiode, laquelle est plus rapide dans le cas (2).
2. a) n(iodure)introduit bécher (1) = 25 × 10–3 × 0,10
= 2,5 × 10–3 mol.
n(iodure)introduit bécher (2) = 25 × 10–3 × 0,20
= 5,0 × 10–3 mol.
n(peroxyde d’hydrogène)introduit = 10 × 10–3 × 0,10
= 1,0 × 10–3 mol.
xmax = 1,0 × 10–3 mol : le peroxyde d’hydrogène est
le réactif limitant dans les deux systèmes chimi-
ques étudiés.
Les ions iodure sont bien en excès, mais l’excès sert
à solubiliser le diiode formé sous forme d’ions triio-
dure I3
–.
b) Le peroxyde d’hydrogène étant le réactif limi-
tant dans les deux systèmes chimiques, il se forme
la même quantité de diiode au final dans les deux
systèmes. L’aspect des deux béchers est identique
en fin de transformation.
3. La concentration de l’un des réactifs a été dou-
blée : c’est le facteur cinétique mis en jeu dans ce
protocole A.
4. La formation de diiode permettant le suivi tem-
porel de la solution, il importe de s’assurer que le
diiode formé soit solubilisé au fur et à mesure de sa
production. Il n’est possible ici de jouer sur la
concentration du réactif peroxyde d’hydrogène
qu’à la condition qu’il reste le réactif limitant et
que les ions iodure soient en excès suffisant.
Autre proposition :
On ne change rien pour l’erlenmeyer (1) :
10 mL d’eau oxygénée acidifiée de concentration
C0 + 25 mL de solution aqueuse d’iodure de potas-
sium, de concentration C2 en soluté apporté.
Pour l’erlenmeyer (2) :
10 mL d’eau oxygénée acidifiée de concentration
C0 + 10 mL d’eau + 25 mL de solution aqueuse
d’iodure de potassium, de concentration C2 en
soluté apporté.
5. Le contenu de l’erlenmeyer (3) présente une
teinte plus foncée que celui de l’erlenmeyer (4) au
•

7
Chapitre 1 - Transformations lentes et rapides
bout de 2 min. Le système chimique (3) évolue donc
plus vite que le système (4).
6. Le facteur cinétique mis en jeu dans ce protocole
est la température du milieu réactionnel.
Suivi optique
d’une dismutation (page 24)
Malgré un temps de réponse voisin du dixième de
seconde, l’œil constitue un outil performant pour
suivre la formation lente de soufre colloïdal au cours
de cette transformation chimique.
1. La formation de soufre colloïdal, S(s), permet un
suivi visuel de la transformation chimique.
2. La formation progressive de soufre colloïdal,
S(s), opacifie la solution. La durée de disparition de
la croix pour l’œil placé au-dessus du bécher peut
constituer un outil de comparaison pour mettre en
évidence les facteurs cinétiques (cf. exercice 21,
page 30 du livre élève).
3. a) S2O3
2– (aq) + 6 H+(aq) + 4 e–
= 2
S(s)
+ 3 H2O(l)
S2O3
2– (aq)
+ H2O(l) = 2 SO2(aq) + 2 H+(aq) + 4 e–.
On retrouve ainsi l’équation de la réaction en som-
mant les deux demi-équations.
b)
Réduction
S2O3
2–(aq) + S2O3
2–(aq) + 4 H+(aq)
2 S(s) + 2 SO2(aq) + 2 H2O.
Oxydation
4. L’ion thiosulfate est à la fois l’oxydant (réduc-
teur conjugué : le soufre) et le réducteur (oxydant
conjugué : le dioxyde de soufre) dans cette réac-
tion d’oxydoréduction.
5. Dans cette réaction d’oxydoréduction, l’ion
thiosulfate s’oxyde et se réduit, ce qui justifie le
terme de dismutation.
Suivi cinétique de transformations
avec solides (page 24)
Cette activité permet d’aborder la cinétique des
réactifs à l’état solide. Nous ne sommes pas dans le
programme, mais bien pour comprendre expéri-
mentalement pourquoi le chimiste, l’industriel pri-
vilégient un tel état de division lorsqu’il utilise des
solides.
1. a) Cu2+(aq) + 2 e– =
Cu(s)
Zn(s) = Zn2+(aq) + 2 e–
.
Les deux demi-équations mettent en évidence un
transfert d’électrons.
A
C
T
I
V
I
T
É
3
A
C
T
I
V
I
T
É
3
A
C
T
I
V
I
T
É
4
A
C
T
I
V
I
T
É
4
b) L’oxydant est l’ion cuivre (II), de couleur bleue
en solution aqueuse.
2. Les ions cuivre (II) étant consommés au cours de
la transformation, la solution contenue dans le
bécher s’éclaircit progressivement.
3. La solution s’éclaircit plus rapidement dans le
bécher contenant le zinc en poudre.
4. La transformation chimique s’effectue plus rapi-
dement lorsque l’état de division du solide est
important. En utilisant de la poudre plutôt que de
la grenaille, on augmente la surface de contact
entre les réactifs : la transformation est plus
rapide.
L’état de division d’un solide modifie la durée d’évo-
lution du système chimique : c’est un facteur ciné-
tique.
Titrage d’une solution aqueuse
de peroxyde d’hydrogène (page 25)
Ce TP ECE consiste à réaliser un titrage d’oxydoré-
duction mettant en jeu la manganimétrie, il permet
de vérifier les savoir-faire expérimentaux concer-
nant :
– la réalisation d’une dilution (choix du matériel,
mode opératoire) ;
– le prélèvement d’une solution (pipetage dans un
bécher, rinçage de la pipette avec la solution à préle-
ver, utilisation d’un pipeteur, repérage correct des
niveaux) ;
– la fiole jaugée (ajustement au trait de jauge et
homogénéisation) ;
– la burette graduée (rinçage de la burette avec la
solution à prélever, ajustement du zéro) ;
– l’agitateur magnétique (position et vitesse cor-
rectes du barreau aimanté) ;
– repérage de l’équivalence.
➜ 1 Transformation étudiée
1. MnO4
–(aq) + 8 H+(aq) + 5 e– = Mn2+(aq) + 4 H2O(l)
H2O2(aq) = O2(aq) + 2 H+(aq) + 2 e–.
2. On retrouve l’équation de la réaction en multi-
pl iant l a première demi-é quat ion par 2 et l a se conde
par 5 :
2 MnO4
–(aq) + 5 H2O2(aq) + 6 H+(aq)
2 Mn2+(aq) + 5 O2(g) + 8 H2O(l).
3. Le peroxyde d’hydrogène est le réducteur (oxy-
dant conjugué : le dioxygène) dans cette réaction
d’oxydoréduction.
A
C
T
I
V
I
T
É
5
A
C
T
I
V
I
T
É
5

8Partie 1 - La transformation d’un système chimique est-elle toujours rapide ?
➜ 2 Dosage de la solution diluée de peroxyde
d’hydrogène
1. Le volume VE versé pour atteindre l’équivalence
est de l’ordre de 22 mL.
2. a) Le réactif limitant juste avant l’équivalence
est l’ion permanganate.
b) À l’équivalence, il y a changement de réactif
limitant : le peroxyde d’hydrogène complètement
consommé dans l’erlenmeyer devient le réactif
limitant.
c) Lorsque l’équivalence est atteinte, la première
goutte de réactif titrant versé colore la solution
contenue en rose violacé.
3. a) C1 · V1 = 2 C2 · VE ; C1 = 2·
2E
1
CV
V.
Avec VE = 22,0 mL, on trouve :
C1 = 8,8 × 10–2 mol · L–1.
b) C = 10 C1 = 8,8 × 10–1 mol · L–1.
Pour retrouver le titre, on multiplie par 11,2 :
8,8 × 10–1 × 11,2 = 9,9 volumes.
Corrigés des exercices (page 26)
1 Définitions
Une réaction d’oxydoréduction correspond à un
transfert d’électrons, alors qu’une réaction acido-
basique correspond à un transfert de protons.
2 Oxydoréduction
1. Un oxydant est une espèce chimique susceptible
de capter un ou plusieurs électrons.
2. Un réducteur est une espèce chimique suscepti-
ble de perdre un ou plusieurs électrons.
3 Vrai ou faux
a) Vrai.
b) Faux : texte à venir
c) Faux : texte à venir
d) Vrai.
4 Écrire une demi-équation
C6H8O6(aq) = C6H6O6(aq) + 2 H+(aq) + 2 e–.
5 Ajuster une demi-équation
Cr2O7
2–(aq) + 14 H+(aq) + 6 e– = 2 Cr3+(aq) + 7 H2O(l).
6 Écrire une réaction d’oxydoréduction
Écrire une réaction d’oxydoréduction
MnO4
–(aq) + 4 H+(aq) + 3 Fe2+(aq)
MnO2(s) + 2 H2O(l) + 3 Fe3+(aq).
7 Vous avez dit titrer ! (vrai ou faux ?)
a) Vrai.
b) Vrai.
c) Faux : avant l’équivalence, le réactif limitant est
l’espèce chimique contenue dans la solution pré-
sente dans le bécher.
d) Vrai.
e) Faux.
8 Rapide ou instantanée ?
1. Sa durée est trop courte pour que l’évolution soit
suivie à l’œil ou avec des appareils de mesure cou-
rants. La durée d’évolution du système est infé-
rieure ou égale à la durée de persistance rétinienne
(1/10 s).
2. Beaucoup de transformations rapides sont
considérées comme instantanées, mais certaines
transformations sont rapides sans être instanta-
nées. On ne dispose pas de moyens pour étudier
leur rapide évolution.
9 Trempe
1. Il s’agit de bloquer une transformation chimique
en jouant sur le facteur cinétique température.
2. Un refroidissement brutal du milieu réactionnel
(avec de la glace, par exemple) fige le système dans
l’état où il se trouve et cela permet donc de l’analy-
ser dans cet état.
10 Transformation ultrarapide
1. L’apport d’énergie du détonateur permet d’en-
clencher la transformation chimique, laquelle est
très rapide.
2. Il faut 130 g d’azoture de sodium.
11 Manganimétrie
1. a) MnO4
–(aq) + 8 H+(aq) + 5 e–
= Mn2+(aq) + 4 H2O(l)
MnO4
–(aq) + 4 H+(aq) + 3 e– = MnO2(s) + 2 H2O(l).
b) En milieu fortement acidifié par de l’acide sulfu-
rique, le couple à prendre en compte est :
MnO4
–(aq)/Mn2+(aq).
c) En milieu fortement acidifié, le réducteur conju-
gué de l’ion permanganate est l’ion manganèse
(II), incolore. Le passage d’une espèce chimique
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
1
/
169
100%