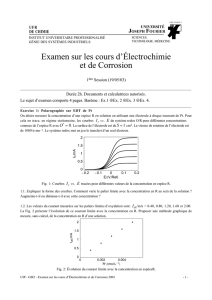
Cours d'électrochimie - L3 Chimie-Physique et M1 Matériaux concours
François Henn
Janvier 2006
1
Plan du cours d’électrochimie
L3 Chimie-Physique et M1 materiaux concours
2006/2007
- I) Introduction et rappels :
Rappel des liens fondamentaux entre électrochimie et thermodynamique: affinité
chimique et électrochimique, degrés d’avancement et vitesse de réaction
- II) Systèmes hors d’équilibre
- Création d’entropie et Surtension , Loi empirique de Tafel, Etude de la vitesse d’un
processus électrochimique (faradique) simple (modèle fondé sur les courbes d’enthalpie
libre), Loi de Butler-Volmer, réaction lente et rapide
- III) Interface Electrode/Solution
Processus non faradique, double couche et couche diffuse, Représentation de l'interface
par un circuit RC équivalent et cinétique de relaxation associée, Réactions contrôlées par
le transfert de masse: courant limite, Spectrométrie d’Impédance Complexe : un outil de
choix pour étudier les phénomènes interfaciaux.
- IV) Electrochimie appliquée I : accumulateurs primaires (piles) et secondaires
(batteries)
L'accumulateur électrochimique: une machine (complexe) pour transformer l'énergie
chimique en énergie électrique, et vice versa, Principe de fonctionnement et architectures,
Caractéristiques et critères de performance, Cas des piles : piles salines et alcalines,
Electrodes à gaz : couples Zn/air et piles à combustible, Cas des
accumulateurs rechargeables: batteries au plomb, Ni(OH)
2
, Li-ions
- V) Electrochimie appliquée II : corrosion
La corrosion : un phénomène thermodynamiquement inévitable (conséquence sur notre
économie), Corrosion sèche, Corrosion humide, Piles de corrosion/corrosion
différentielles, Facteurs thermodynamiques (diagramme potentiel/pH –Pourbaix),
Facteurs cinétiques (diagrammes d’Evans)

Cours d'électrochimie - L3 Chimie-Physique et M1 Matériaux concours
François Henn
Janvier 2006
2
Chapitre I:
Introduction et Rappels
A) Introduction Générale
On aborde généralement l’électrochimie en associant réaction d'oxydo-reduction et
thermodynamique des états d'équilibre. Cette approche conduit à la loi de Nernst qui relie le
potentiel d'une électrode à la concentration des espèces impliquées. (nb: c'est une façon de
mesurer des constantes d'équilibre!). On applique alors cette électrochimie des états
d'équilibre à l’étude des potentiels d’électrode puis à l’analyse des solutions ioniques diluées
(et aux différentes méthodes de titration). Si ces aspects sont très importants, ils ne
représentent qu’une partie de l’électrochimie; cette science va bien au-delà. Voici quelques
exemples :
- études des sels fondus (milieux ioniques en fusion) et/ou des milieux fortement
concentré: étude des systèmes réels (mesure des coefficients d'activité)
- réactivité en milieux aqueux/non aqueux : catalyse, synthèse organique et toute
réaction faisant intervenir des transferts d’électrons
- application aux solides :
- 1) corrosion (nouveaux alliages dans l’aéronautique, l’automobile, le
bâtiment, l’électronique,..…..) (sera vue au chapitre V)
- 2) accumulation électrochimique de l’énergie (piles et batteries):
développement récent miniaturisation (éléments portables), véhicules
électriques ou hybrides (sera vue au chapitre IV)
- 3) capteur (miniaturisation : micro-électrodes pour application médicale ou
environnementale –détection de certains ions dans les eaux, les sols….)
Toutes ces applications mettent en jeu des réactions d’oxydoréduction, c’est à dire de transfert
électronique entre composés d'affinité électronique (ou d'électronégativité) différente. On peut
donc appliquer toutes les notions thermodynamiques connues à ce type de réaction chimique :
définition des conditions d’équilibre, loi d’action de masse, ..etc.. Nous comprenons très
facilement, à travers les exemples suscités, que l’électrochimie est une science
pluridisciplinaire qui nécessite également des compétences en : cinétique, électricité, science
des interfaces, phénomène de transport de matière, mécanismes réactionnels (transfert
d’électron)….Notons aussi que le développement récent des nanotechnologies joue un rôle
important en électrochimie (nano-ressource d’énergie, nano-capteur(électrodes), nano-
réacteurs…)
Arrêtons-nous, dans un premier instant, sur l’aspect thermodynamique (que vous connaissez
mieux). Vous avez étudié l’électrochimie des états d’équilibre (ex loi de Nernst et diagramme
Potentiel/pH). Mais pour comprendre les phénomènes qui ont cours dans la plupart des
domaines d’application de l’électrochimie, cela ne suffit pas. Tant que l’on mesure des
retour plan

Cours d'électrochimie - L3 Chimie-Physique et M1 Matériaux concours
François Henn
Janvier 2006
3
échanges d’électrons (c’est à dire du courant = flux de charges), cela signifie que des
réactions ont lieu et que l’on est par conséquent dans des conditions hors d’équilibre.
La notion de potentiel d’électrode est intimement liée à la notion d’équilibre. Les conditions
hors d’équilibre se caractérisent (2
ème
principes de la thermo) par la production constante
d’entropie interne. En électrochimie, cette croissance d’entropie est caractérisée par des
surtensions, c'est à dire par l'écart qui apparaît entre le potentiel au repos (i=0) donné par la
loi de Nernst et le potentiel de fonctionnement (i≠0). Nous reviendrons en détails sur cette
notion fondamentale de surtension.
Une partie importante de l'électrochimie fondamentale essaie de modéliser le phénomène de
surtension. Nous verrons dans la suite de ce cours que cela fait intervenir un grand nombre de
processus différents et complexes qui se superposent les uns aux autres: cinétique de transfert
électronique, effet joule, transfert de matière, effets non faradiques (capacitif). La plupart de
ces phénomènes ne sont pas linéaires ; c'est-à-dire qu’ils ne dépendent pas linéairement du
courant qui circule dans la cellule électrochimique.
B) Rappels : liens avec la thermodynamique des états d’équilibres
Electrochimie : Etude des réactions d’oxydo-réduction lorsqu'il y a échange de charges
électroniques à l’interface d'une électrode
1
Red Ox +nF e
-
On peut considérer l’électron comme une espèce chimique et exprimer alors le degrés
d’avancement ξ à partir des relations déjà définies en thermodynamique:
nF
dn
d
e
=ξ
où n
e
est le nombre d’électrons échangé par unité de surface, F ( Faraday : charge d’une mole
d’électron) et n le coefficient stœchiométrique associée à cette espèce.
Alors la vitesse de réaction v devient :
nF
i
nF
dt
dn
dt
d
v
e
=== 1
.
ξ
et
ξ
dnFdti ..
=
i est une densité de courant (par unité de surface A/m
2
). Le courant passant dans le circuit est
donc directement lié à la vitesse de la réaction. Notons aussi que i et dξ sont de même signe.
1
Si l'échange de charge électronique s'effectue directement d'une espèce à l'autre au sein d'une solution
(liquide ou solide), la reaction d'oxydo-reduction est alors traitée comme toute réaction chimique (ex
acide/base) et le recours à la mesure des potentiels n'est pas necessaire (autant passer par le ∆
∆∆
∆G de la
réaction et les constantes d'équilibre)

Cours d'électrochimie - L3 Chimie-Physique et M1 Matériaux concours
François Henn
Janvier 2006
4
Affinité Electrochimique d’une cellule galvanique
2
Schéma : Métal/matière active/électrolyte//électrolyte/matière active/Métal
Cas de la pile Daniell : Zn/Zn
2+
//Cu
2+
/Cu/Zn
(avec E
0
(Zn/Zn
2+
)= -0.76eV E
0
(Cu/Cu
2+
)=0.34V)
Naturellement le système évoluera dans le sens d'une oxydation du zinc et d'une réduction du
cuivre. (
Rappel: le courant circule dans le sens des cations et dans le sens opposé a celui des électrons)
Zn/Zn
2+
constitue donc l'anode et Cu/Cu
2+
la cathode
L’énergie interne U d’une telle cellule (plusieurs phases en contact) peut être définie de la
manière suivante :
dtiEEpdVdQdtiEdWdQdU
ac
ee
)( −+−=∆++=
où
ac
E
E
−
est la tension ∆E au borne de la cellule et
e
dQ la chaleur échangée.
Le système est hors d’équilibre : QdQdTdS
ie
+= d’où QdTdsQd
ie
−=
Sachant que la fonction enthalpie libre G=U+PV+TS, on aboutit à :
dtiEQdVdpsdTdG
i
∆+−+−=
ou
ξ
dnFEQdVdpsdTdG
i
∆+−+−=
qui finalement en conditions isobare et isotherme se réduit à :
ξ
dnFEQddG
i
∆+−=
or nous savons que A (l’affinité) est égale à
T,p
)
G
(ξ∂
∂
−
.
Si l’on divise l’expression de dG par dξ, on arrive à :
nFE
d
dG
dQd
i
∆+−=
ξξ
on en déduira l’affinité électrochimique à comme :
nFEA
dQd
A
i
∆+==
ξ
~
à joue le même rôle que A (cas d’un système purement chimique)
Les conditions de repos (i=0) (partie électrochimique de la réaction) imposent Ã=0 soit :
)(
r
r
GnFEEnFA ∆=∆−=∆−=
2
Il existe 2 types de cellules électrochimiques (constituées de 2 couples redox) : galvaniques et électrolytiques
1) Galvanique : la réaction entre les 2 systèmes est spontanée : cas des piles primaires, piles à
combustible, des protections galvaniques (anti-corrosion)
2) Electrolytique : le courant passe dans le circuit parce qu’on applique un potentiel aux électrodes (on
place le système de manière à ce qu’il s’écarte de l’équilibre). Electrolyse de l’eau, dépôt de
revêtement métallique
Nb : les piles secondaires (ou rechargeables) se comportent comme des cellules galvaniques en décharge (on
récupère spontanément l’énergie du système) ou comme des cellules électrolytiques lorsqu’on les recharge

Cours d'électrochimie - L3 Chimie-Physique et M1 Matériaux concours
François Henn
Janvier 2006
5
où
r
E∆
est le potentiel aux bornes du système lorsqu'il est au repos i=0 . Il en découle
directement :
nF
A
EEE
a
r
c
rr
−=−=∆ (cathode/anode)
3
avec
a
r
E potentiel de l'anode (siège de l'oxydation) et
c
r
E potentiel de la cathode (siège de la
réduction)
On peut ainsi écrire que l’affinité A du système est la somme des affinités de 2 sous systèmes
correspondant respectivement à l’anode et la cathode tel que A=A
c
+A
a
C’est de cette manière que l’on relie l’évolution du système à la différence de potentiel entre
les deux couples et que l’on peut calculer le potentiel d’équilibre (ou de repos - en circuit
ouvert-) Loi de Nernst.
retour plan
3
C'est une autre forme de la relation de Nernst
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
1
/
47
100%