Chimie PCSI: Configurations électroniques, équilibres, thermodynamique
Telechargé par
Ah Med

Kevin Moris
Professeur de Chimie en PCSI
au lycée Vaugelas (Chambéry)
Philippe Hermann
Professeur de Chimie en PCSI
au lycée Victor Hugo (Besançon)
Yves Le Gall
Professeur de Chimie en PCSI
au lycée Lakanal (Sceaux)
9782100556267-Moris-lim.qxd 12/05/11 15:01 Page I


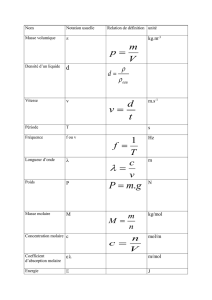
1• La cinématique
III
© Dunod. La photocopie non autorisée est un délit.
Chapitre 1 • Les lois de Newton
Table des matières
1. Configurations électroniques 1
1.1 Structure d’un atome 1
1.2 Cas de l’atome d’hydrogène 2
1.3 Les nombres quantiques 2
1.4 Atomes polyélectroniques 3
1.5 Schéma de Lewis des atomes 6
1.6 Cas des ions 6
Synthèse 7
Tests et exercices 8
Corrigés des exercices 10
2. Classification périodique
des éléments 13
2.1 Présentation rapide 14
2.2 Évolution de quelques propriétés atomiques 15
Synthèse 18
Tests et exercices 19
Corrigés des exercices 22
3. Schémas de Lewis
des molécules. Règles VSEPR 24
3.1 La liaison chimique selon Lewis 24
3.2 Représentation de Lewis 26
3.3 Prévision de la géométrie de molécules 31
Synthèse 34
Tests et exercices 35
Corrigés des exercices 38
4. La cinétique du point de vue
macroscopique 43
4.1 Vitesses en cinétique chimique 44
4.2 Facteurs influençant la vitesse de réaction 48
4.3 Lois de vitesse et cinétique formelle 53
4.4 Détermination expérimentale des ordres 59
Synthèse 65
Tests et exercices 67
Corrigés des exercices 73
5. La cinétique du point de vue
microscopique 83
5.1 Actes élémentaires 83
5.2 Mécanismes réactionnels 91
Synthèse 101
Tests et exercices 102
Corrigés des exercices 108
6. Équilibres acido-basiques.
Détermination de l’état d’équilibre
d’une solution aqueuse 117
6.1 Définitions préliminaires 117
6.2 Constante d’équilibre : Ko
T120
6.3 Quotient de réaction et constante
d’équilibre 122
6.4 Réactions acido-basiques 122
6.5 Force d’un acide ou d’une base 123
9782100556267-Moris-tdm.qxd 12/05/11 15:19 Page III
www.biblio-scientifique.net

IV
Table des matières
6.6 Couples acido-basiques de l’eau –
Effet nivelant du solvant 124
6.7 Diagrammes de prédominance
et de distribution 125
6.8 Prévision du sens d’échange du proton 127
6.9 Calculs de pH 130
6.10 Application à un dosage acido-basique 141
Synthèse 144
Tests et exercices 146
Corrigés des exercices 150
7. Les réactions
de complexation 156
7.1 Échange de cations et d’anions 156
7.2 Constantes d’équilibre 157
7.3 Diagramme de prédominance –
Diagramme de distribution 159
7.4 Prévision du sens d’échange de la particule 161
7.5 Détermination de l’état d’équilibre
d’une solution aqueuse 162
7.6 Notion de réaction prépondérante
généralisée 166
7.7 Titrage complexométrique 168
Synthèse 170
Tests et exercices 171
Corrigés des exercices 176
8. Les réactions
de précipitation 184
8.1 Échange de cations et d’anions 184
8.2 Équilibre solide / espèces en solution –
constante d’équilibre 185
8.3 Effet d’ion commun –
Influence sur la solubilité 189
8.4 Dosage par précipitation 190
Synthèse 192
Tests et exercices 193
Corrigés des exercices 196
9. Les réactions
d’oxydo-réduction 204
9.1 Échange d’électrons 204
9.2 Définitions fondamentales 205
9.3 Nombres d’oxydation 205
9.4 Cellule électrochimique 210
9.5 Pile électrochimique 210
9.6 Potentiel d’électrode ou potentiel
d’oxydoréduction 211
9.7 Relation de Nernst 212
9.8 Prévision des réactions rédox 212
9.9 Méthode de la réaction prépondérante 214
9.10 Calcul d’un potentiel standard inconnu 215
9.11 Dosage rédox 217
Synthèse 219
Tests et exercices 220
Corrigés des exercices 224
10. Thermodynamique
des systèmes chimiques 230
10.1 Modèles d’étude des transformations 231
10.2 États standard, grandeurs molaires standard 234
10.3 Grandeurs standard de réaction 237
10.4 Application :
étude des transferts thermiques 243
10.5 Formation d’un constituant
physico-chimique 247
10.6 Thermochimie de quelques transformations 251
Synthèse 254
Tests et exercices 255
Corrigés des exercices 261
11. Éléments de cristallographie
(Option SI) 269
11.1 L’état solide cristallin 269
11.2 Définitions de base 270
11.3 Cristaux métalliques 272
11.4 Les cristaux ioniques 281
9782100556267-Moris-tdm.qxd 12/05/11 15:19 Page IV
www.biblio-scientifique.net

V
© Dunod. La photocopie non autorisée est un délit.
Table des matières
11.5 Les cristaux covalents 287
11.6 Les cristaux moléculaires 289
Synthèse 291
Tests et exercices 292
Corrigés des exercices 296
Fiches méthode 301
1. Comment écrire un mécanisme réactionnel
en chimie organique ? 301
2. Méthode de la réaction prépondérante 303
3. Comment bien travailler en prépa ? 304
4. Comment bien rédiger une copie ? 306
Classification périodique
des éléments 308
Index 309
9782100556267-Moris-tdm.qxd 12/05/11 15:19 Page V
www.biblio-scientifique.net
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
1
/
320
100%