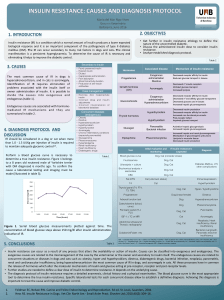
Closed-Loop Control of Blood Glucose: Lecture Notes
Telechargé par
EL HACHIMI Mohamed


Lecture Notes
in Control and Information Sciences 368
Editors: M. Thoma, M. Morari

Frederick Chee, Tyrone Fernando
Closed-Loop Control of Blood
Glucose
ABC

Series Advisory Board
F. Allgöwer, P. Fleming, P. Kokotovic,
A.B. Kurzhanski, H. Kwakernaak,
A. Rantzer, J.N. Tsitsiklis
Authors
Frederick Chee
School of Electrical
Electronic and Computer Engineering
University of Western Australia
35 Stirling Highway
Crawley, WA 6009
Australia
Email: hf.[email protected]
Tyrone Fernando
School of Electrical
Electronic and Computer Engineering
University of Western Australia
35 Stirling Highway
Crawley, WA 6009
Australia
Email: t[email protected]
Library of Congress Control Number: 2007932674
ISSN print edition: 0170-8643
ISSN electronic edition: 1610-7411
ISBN-10 3-540-74030-9 Springer Berlin Heidelberg New York
ISBN-13 978-3-540-74030-8 Springer Berlin Heidelberg New York
This work is subject to copyright. All rights are reserved, whether the whole or part of the material is
concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting,
reproduction on microfilm or in any other way, and storage in data banks. Duplication of this publication or
parts thereof is permitted only under the provisions of the German Copyright Law of September 9, 1965, in
its current version, and permission for use must always be obtained from Springer. Violations are liable for
prosecution under the German Copyright Law.
Springer is a part of Springer Science+Business Media
springer.com
c
Springer-Verlag Berlin Heidelberg 2007
The use of general descriptive names, registered names, trademarks, etc. in this publication does not imply,
even in the absence of a specific statement, that such names are exempt from the relevant protective laws and
regulations and therefore free for general use.
Typesetting: by the authors and SPS using a Springer L
A
T
E
X macro package
Printed on acid-free paper SPIN: 12077946 89/SPS 5 43210

To Jehovah God, my parents and siblings
Frederick
To my wife Yasmin and two daughters Melissa and Rochelle
Tyrone
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
1
/
166
100%