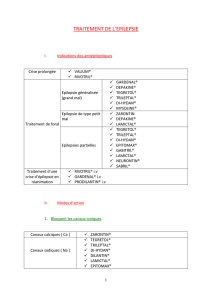
Porphyries: Maladies génétiques et approches thérapeutiques

Les porphyries
Unité d’Enseignement:
Mécanismes de survenue des maladies génétiques et
approches thérapeutiques
Université Cheikh Anta Diop
FMPO, Laboratoire de Biochimie Pharmaceutique Année Universitaire 2018 - 2019
Master I/ Semestre 2

Plan
I) Introduction
II) Eléments de biosynthèse de l’hème
III) Classification
IV) Description clinique
V.1Porphyries érythropoiëtiques
V.2 Porphyries érythropoëtiques
VI Diagnostic Biologique
Conclusion

Définition : grpes affections dues à des anomalies
enzymatiques de la voie de la synthèse de l’héme.
Ksées/accumulation et excrétion des porphyrines et de
leurs précurseurs (acide δ-aminolévulinique et
porphobilinogène).
Le déficit enzymatique est incomplet, Manifestation que
lorsque les besoins hépatiques en hème sont augmentés.
On distingue les porphyries acquises et les porphyries
héréditaires.
I) Introduction

I) Introduction
Porphyries héréditaires : déficit d’une des enzymes de
la synthèse de l’ hème,
mutations d’un gène, transmission très souvent
autosomique dominante.
La porphyrie aigüe intermittente = plus fréquente
des porphyries héréditaires.
Porphyries acquises :
la porphyrie cutanée sporadique (ou tardive) : la
plus fréquente des porphyries et est acquise dans 80%
des cas.
Les formes acquises souvent associées à une atteinte
hépatique.

(1): ALA-synthase; (2): PBG-synthase (ALA déhydratase);(3) : PBG-déaminase; (4): Uroporphyrinogène III synthase;
(5): Uroporphyrinogène décarboxylase; (6):Coproporphyrinogène oxydase; (7):Protoporphyrinogène oxydase;
(8):Ferrochélatase
II) Eléments de biosynthèse de l’hème
7 réactions: 8 enzymes
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
1
/
43
100%