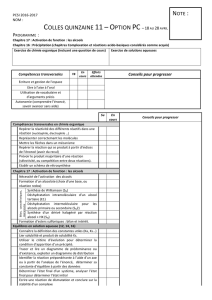
CHIMIE ORGANIQUE 1 A. ATMANI 2013-2014 UNIVERSITE DE TLEMCEN

CHIMIE ORGANIQUE 1
S5-Licence Chimie
CHIMIE ORGANIQUE 1
A. ATMANI
2013-2014
UNIVERSITE DE TLEMCEN
U.TLEMCEN
1

http://www.science.gouv.fr
Non-
métaux
Métaux
alcalino-
terreux
Métaux
pauvres Gaz rares
Lanthanides
Métaux
alcalins
Métaux de
transition HalogènesActinides
Élément
gazeux**
Élément
artifi ciel Uut*
Élément
liquide**
Famille
** Dans des conditions normales
de température et de pression
0oC, 1 atm
* non reconnu à ce jour
par l’UICPA
Élément
solide
opixido
Élément
artifi ciel
Hydrogène
Francium
Césium
Rubidium
Potassium
Sodium
Lithium
Hélium
UnunoctiumUnunseptium
Radon
Xénon
Krypton
Argon
Néon
Astate
Iode
Brome
Chlore
Fluor
Ununhexium
Polonium
Tellure
Sélénium
Soufre
Oxygène
Ununpentium
Bismuth
Antimoine
Arsenic
Phosphore
Azote
Ununquadium
Plomb
Étain
Germanium
Silicium
Carbone
Ununtrium
Thallium
Indium
Gallium
Aluminium
Bore
Copernicium
Mercure
Cadmium
Zinc
Roentgenium
Or
Argent
Cuivre
Darmstadtium
Platine
Palladium
Nickel
Meitnerium
Iridium
Rhodium
Cobalt
Hassium
Osmium
Ruthénium
Fer
Bohrium
Rhénium
Technétium
Manganèse
Seaborgium
Tungstène
Molybdène
Chrome
Dubnium
Tantale
Niobium
Vanadium
Rutherfordium
Hafnium
Zirconium
Titane
Actinium
Lanthane
Yttrium
Scandium
Radium
Baryum
Strontium
Calcium
Magnésium
Béryllium
Lawrencium
Lutétium
Nobélium
Ytterbium
Mendélévium
Thulium
Fermium
Erbium
Einsteinium
Holmium
Californium
Dysprosium
Berkélium
Terbium
Curium
Gadolinium
Américium
Europium
Plutonium
Samarium
Neptunium
Prométhium
Uranium
Néodyme
Protactinium
Praséodyme
Thorium
Cérium
Fr
Cs
Rb
K
Na
Li
He
Uuo
Rn
Xe
Kr
Ar
Ne
At
Uus*
I
Br
Cl
F
Uuh
Po
Te
Se
S
O
Uup*
Bi
Sb
As
P
N
Uuq
Pb
Sn
Ge
Si
C
Uut*
Tl
In
Ga
Al
B
Cn
Hg
Cd
Zn
Rg
Au
Ag
Cu
Ds
Pt
Pd
Ni
Mt
Ir
Rh
Co
Hs
Os
Ru
Fe
Bh
Re
Tc
Mn
Sg
W
Mo
Cr
Db
Ta
Nb
V
Rf
Hf
Zr
Ti
Ac
La
Y
Sc
Ra
Ba
Sr
Ca
Mg
Be
Lw
Lu
No
Yb
Md
Tm
Fm
Er
Es
Ho
Cf
Dy
Bk
Tb
Cm
Gd
Am
Eu
Pu
Sm
Np
Pm
U
Nd
Pa
Pr
Th
Ce
H
1
87
55
37
19
11
3
2
118117
86
54
36
18
10
85
53
35
17
9
116
84
52
34
16
8
115
83
51
33
15
7
114
82
50
32
14
6
113
81
49
31
13
5
112
80
48
30
111
79
47
29
110
78
46
28
109
77
45
27
108
76
44
26
107
75
43
25
106
74
42
24
105
73
41
23
104
72
40
22
89
57
39
21
88
56
38
20
12
4
103
71
102
70
101
69
100
68
99
67
98
66
97
65
96
64
95
63
94
62
93
61
92
60
91
59
90
58
Nom
X
Numéro atomique
Symbole atomique
MMasse molaire atomique
(g.mol-1)
Famille
Z
1,0
(223)
132,9
85,5
39,1
23,0
6,9
4,0
(294)
(222)
131,3
83,8
39,9
20,2
(210)
126,9
79,9
35,5
19,0
(292) (292)
(209)
127,6
79,0
32,1
16,0
(288)
209,0
121,8
74,9
31,0
14,0
(289)
207,2
118,7
72,6
28,1
12,0
(284)
204,4
114,8
69,7
27,0
10,8
(277)
200,6
112,4
65,4
(272)
197,0
107,9
63,5
(272)
195,1
106,4
58,7
(268)
192,2
102,9
58,9
(269)
190,2
101,1
55,8
(264)
186,2
(98)
54,9
(266)
183,8
95,9
52,0
(262)
180,9
92,9
50,9
(261)
178,5
91,2
47,9
(227)
138,9
88,9
45,0
(226)
137,3
87,6
40,1
24,3
9,0
(260)
175,0
(259)
173,0
(258)
168,9
(257)
167,3
(254)
164,9
(251)
162,5
(247)
158,9
(247)
157,4
(243)
152,0
(244)
150,4
(237)
(145)
238,0
144,2
231,0
140,9
232,0
140,1
118171615141312111098765432
Colonne
1
7
6
5
4
3
2
7
6
Dmitri Ivanovitch Mendeleïev
(1834 – 1907) est un chimiste
russe connu pour ses travaux
sur la classifi cation périodique
des éléments. En 1869, il
publia une première version
de son tableau périodique des
éléments appelé aussi tableau
de Mendeleïev. Il déclara que
les éléments chimiques pouvaient être arrangés selon
un modèle qui permettait de prévoir les propriétés
des éléments non encore découverts.
TABLEAU PÉRIODIQUE DES ÉLÉMENTS
Tableau_Mendeleiev_2011.indd 1 2/01/12 17:16:00

Table des matières
Table des matières i
1 LES ALCANES 1
1.1 Nomenclature .................................... 2
1.2 Isoméries ....................................... 4
1.3 Propriétés physiques ................................. 5
1.4 Propriétés spectrales ................................. 6
1.5 Réactions chimiques des alcanes ........................... 18
1.6 Synthèse des alcanes et des cycloalcanes ...................... 27
1.7 Exemples d’alcanes ................................. 28
2 LES ALCÈNES 33
2.1 Nomenclature .................................... 34
2.2 Structure et propriétés physiques ........................... 34
2.3 Propriétés spectrales ................................. 36
2.4 Préparation des alcènes ................................ 43
2.5 Réactivité et stabilité ................................. 46
2.6 Réactions d’oxydations ............................... 66
2.7 Substitution en α................................... 70
3 LES ALCYNES 77
3.1 Nomenclature .................................... 77
3.2 Propriétés physiques et spectroscopiques ...................... 78
3.3 Acidité des alcynes terminaux ............................ 83
3.4 Réactions des alcynes ................................ 84
3.5 Exemples d’alcynes importants ........................... 91
4 LES DIÈNES 96
4.1 Introduction ..................................... 96
4.2 Spectroscopie dans l’ultra-violet et le visible .................... 97
4.3 Addition 1,2 et 1,4 ..................................104
4.4 Réactions de Diels-Alder ...............................106
4.5 Étude stéréochimique ................................110
5 LES DÉRIVÉS AROMATIQUES 117
5.1 Structure du benzène : Formule de Kékulé .....................117
5.2 Nomenclature ....................................118
5.3 Propriétés physiques et spectrales ..........................120
i

ii TABLE DES MATIÈRES
5.4 Aromaticité et propriété chimique ..........................126
5.5 Réactions de substitution électrophile en série aromatique .............129
5.6 Activation et désactivation du cycle benzénique ...................137
5.7 Réactions de la chaîne latérale des alkylbenzènes ..................139
5.8 Alcénylbenzènes ................................... 141
5.9 Applications en synthèse ...............................143
6 LES HALOGÉNOALCANES 149
6.1 Nomenclature ....................................149
6.2 Propriétés physiques et spectrales ..........................150
6.3 Propriétés spectroscopiques .............................152
6.4 Nucléophilie et électrophilie .............................155
6.5 Réactions de substitution nucléophile sur carbone saturé ..............156
6.6 Interconversion des groupes fonctionnels par des réactions de SN2 .........165
6.7 Réactions d’éliminations des halogénoalcanes ...................168
6.8 Substitution ou élimination .............................. 171
6.9 Organigramme des transformations .........................173
6.10 Halogénoalcanes importants .............................174
7 LES ORGANOMÉTALLIQUES 179
7.1 Introduction .....................................179
7.2 Préparation des composés organométalliques ....................180
8 LES ALCOOLS ET LES PHÉNOLS 189
8.1 Présentation et nomenclature .............................189
8.2 Propriétés physiques et spectrales ..........................190
8.3 Propriétés chimiques des alcools ...........................194
8.4 Élimination sur les alcools ..............................203
8.5 Oxydation ......................................204
8.6 Réaction des éthers et époxydes ........................... 207
8.7 Quelques exemples d’alcools importants ......................209

Chapitre
8LES ALCOOLS ET LES PHÉNOLS
8.1 Présentation et nomenclature . . . . . . . . . . . . . . . . . . . . . . . . 189
8.2 Propriétés physiques et spectrales . . . . . . . . . . . . . . . . . . . . . 190
8.3 Propriétés chimiques des alcools . . . . . . . . . . . . . . . . . . . . . . 194
8.4 Élimination sur les alcools . . . . . . . . . . . . . . . . . . . . . . . . . 203
8.5 Oxydation .................................204
8.6 Réaction des éthers et époxydes . . . . . . . . . . . . . . . . . . . . . . 207
8.7 Quelques exemples d’alcools importants . . . . . . . . . . . . . . . . . 209
Figure 8.1 – Le méthanol
8.1 Présentation et nomenclature
U
n alcool est un composé possédant un groupement hydroxyle
−OH
lié à un atome de carbone
saturé, hybridé
sp3
; un phénol possède un groupement hydroxyle lié à un cycle aromatique.
Figure 8.2 – Le groupement fonctionnel des alcools
On distingue parmi les alcools trois classes, alcools primaires, secondaires et tertiaires.
189
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
1
/
26
100%