Métabolisme des acides aminés et de l`azote (2ème partie)

- 1 -
UE 3 – Biochimie clinique, Nutrition, Métabolisme
Dr Gonthier
Date : 15/09/2016 Plage horaire : 8H30 11H30
Promo : DCEM1 Enseignant : Dr M-P. Gonthier
Ronéistes :
ROBERT Anne
Métabolisme des acides aminés et de l’azote (2ème partie)
I) Introduction
II) Digestion des protéines alimentaires
III) Catabolisme des acides aminés (début de la ronéo)
1) Catabolisme de la fonction amine
2) Catabolisme du squelette carboné
3) Enzymes clés du métabolisme des AA
4) Echange inter-organe en situation de jeûne (couple glucose/alanine)
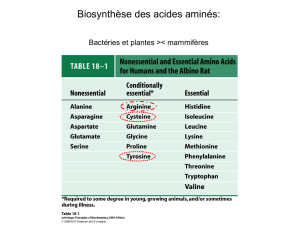
IV) Biosynthèse des acides aminés et dérivés
1) Biosynthèse des acides aminés non essentiels
2) Biosynthèse des dérivés d’acides amines
A. Synthèse de la porphyrine
B. Biosynthèse de la créatinine à partir de glycine et arginine
C. Biosynthèse du glutathion par condensation du glutamate, cystéine et glycine
D. Conversion des acides aminés en amines biologiques par décarboxylation –
synthèse de NO
V) Pathologies associées au métabolisme des acides aminés et de
l’azote
VI) Exploration du métabolisme azoté

- 2 -
III) Catabolisme des acides aminés
Les AA arrivent dans les cellules, qui ont des besoins énergétiques. Les acides aminés vont être catabolisés
afin de refournir de l’énergie sous forme d'ATP. Il existe différentes possibilités pour le catabolisme des
acides aminés, qui implique 2 parties importantes (puisqu’un acide aminé est un acide carboxylique α-
aminé).
De plus, au moment du catabolisme des acides aminés, il existe un grand danger pour l’organisme à cause de
la fonction amine. Il va donc y avoir un traitement spécial de la fonction amine et un traitement spécial pour
le reste de la chaine carbonée.
1) Catabolisme de la fonction amine
Selon la nature du squelette carboné, plusieurs devenirs en fonction des situations physiologiques :
• Lorsque l’on est loin de la situation postprandiale et que l’on a besoin de glucose, la néoglucogenèse
est sollicitée pour cataboliser les acides aminés en vue de former du glucose.
• Si la néoglucogenèse ne suffit plus, il y a formation de corps cétoniques via la cétogenèse.
• Il peut également y avoir la formation d’acides gras grâce à la lipogenèse par la voie du Malonyl-
CoA.
• De plus, en situation où l’on a un besoin immédiat d’énergie il peut y avoir une oxydation directe,
une production d’énergie par le biais du cycle de Krebs et de la chaine respiratoire.
Zoom sur une liaison peptidique entre 2 acides aminés
L’AA1 est couplé au 2ème AA par une liaison peptidique qui peut être rompue par une peptidase. On peut
donc récupérer de l’AA1 ou 2, et chacun aura un devenir particulier concernant la chaîne carbonée en
fonction de la nature du radical, mais les 2 fonctions amines, elles, auront le même devenir.
Après séparation des 2 acides aminés par la peptidase, les 2 fonctions amines, éliminées car la cellule n’a
besoin que du squelette carboné pour générer de l’énergie, sont immédiatement prises en charge. Ces
dernières n’étant pas stables, elles donneront du NH3+ extrêmement toxique et représentent un danger
permanent pour les cellules.
En effet, l’ammoniac est un fort perturbateur du gradient de protons et des électrons de la chaîne respiratoire
ce qui est délétère car nuit au bon fonctionnement de l’ATP synthase.

- 3 -
Rappel : lors de la respiration cellulaire, le transport d’électrons génère un flux de protons qui seront
utilisés par l’ATP synthase. Si l’on ne neutralise pas l’ammoniac, NH₄⁺ sera un puissant découplant qui va
shunter la chaîne respiratoire en captant les protons, ce qui empêchera l’ATP synthase de fonctionner.
Les cellules finissent par mourir, pas forcément par toxicité de NH4+ mais surtout parce qu'elles sont en
manque d'ATP.
L'organisme, au cours de l'évolution a acquis la capacité de se débarrasser de cette fonction NH3 qui est
obligatoirement libérée lors du catabolisme via le cycle de l'urée.
è 95% du mode d'élimination de l'ammoniac.
ATTENTION QCM :
Le cycle de l’urée ou uréogénèse est un mécanisme strictement hépatique.
Il ne se fait pas au niveau des reins.
Le rein intervient uniquement dans l’élimination de l’urée.
Lorsque les tissus périphériques dégradent les AA, la fonction NH3 (pour sortir des tissus périphériques en
l'occurrence et remonter au foie pour générer ce cycle) va être prise en charge par le glutamate, générant de
la glutamine qui est le transporteur de l'ammoniac NH3 au niveau sanguin. Il est ensuite ramené au foie où
la glutamine va être débarrassée de NH3+ pour redevenir du glutamate.
En d'autres termes, la glutamine va prendre en charge ce NH3 pour l’envoyer au niveau de deux organes
clés :
Ø Le foie
La glutamine est à nouveau désaminée pour libérer du NH3 et du glutamate.
Le NH3 libéré est associé à une molécule de CO2 et à de l’ATP pour générer du carbomoyl-phosphate qui
va être condensé par l’ornithine (dérivé de l’arginine) pour former de la citrulline.
La citrulline va elle-même être condensée avec de l’aspartate fournie par le cycle de Krebs et donner de
l’arginosuccinate. Celui-ci est clivé par une lyase en fumarate qui réalimente le cycle de Krebs.
Finalement, l’arginine est désaminée par une arginase et donne la molécule d’urée éliminée dans les urines.
On a affaire à deux enzymes mithochondriales, le reste est cytosolique.

- 4 -
Ø Les reins
La glutamine va également redonner du glutamate (qui repart dans la circulation) et du NH3, converti en
ammonium NH4+ qui lui même est converti en chlorure d'ammonium (Cl-NH4+) et sécrété. C'est ce qu'on
appelle l'ammoniogénèse rénale. (Mais toutes les cellules possédant du NH3 et des H+ sont capables de faire
de l'ammoniogénèse ! )
Remarque : les personnes qui sont en situation de régime hyper protéiné présentent au bout de quelques
années une insuffisance rénale dûe à un métabolisme extrêmement important du cycle de l'urée. Toutes ces
enzymes sont des enzymes Michaeliennes. S'il y a trop de substrat, elles sont ralenties. On finit par ne plus
assurer un cycle normal de l'urée et de l'ammoniogénèse ce qui explique l'altération de la fonction rénale.
De plusi, le cycle de Krebs est très altéré chez les personnes présentant des cirrhoses et chez les personnes
ayant subies une hépatite. C'est pour cela que les médecins demandent à ces patients de contrôler leur
apport en protéines pour éviter une arrivée massive d'AA.
Les AA, contrairement aux glucose et aux AG n'ont pas de " stock" en soi. Quand on consomme des AA, le
foie les utilise pour ses propres besoins, ensuite ces AA sont transmis aux autres tissus et c'est le muscle qui
est capable de garder un peu de protéines en plus. Mais il n'y a pas de stock mirobolant d'AA sauf si vous
êtes en situation de régime hyper protéiné et dans ce cas, une voie musculaire spécifique se met en place.
2) Catabolisme du squelette carboné
Alors en terme de devenir, le reste de la chaîne carbonée va subir différentes possibilités de dégradation
selon la nature de l'AA. Il va y avoir des voies communes et en l'occurrence, selon les groupes et le type, le
catabolisme de la chaine carbonée va aboutir sur une production de substrats intermédiaires associés au
cycle de Krebs :

- 5 -
Ce cycle permet donc à la cellule de tirer profit pour générer de l'ATP en situation postprandiale. Quand on
va s'éloigner de la période post prandiale, on est en situation de jeûne et il faut absolument qu'il y ait de la
néoglucogénèse.
En venant alimenter ce cycle, les AA vont surtout générer une quantité importante d'oxaloacétate permettant
ainsi de remonter à la voie de synthèse du glucose et également du pyruvate.
Lorsque le jeûne se prolonge, il y a possibilité pour les AA de contribuer à la synthèse des corps cétoniques
via la production importante d'Acétyl CoA, qui va générer une quantité importante d'acétoacétate. Ce dernier
va générer du béta-hydroxybutyrate et de l'acétone (élimination pulmonaire).
Ces corps cétoniques vont alimenter le cerveau (entre autres) pour reproduire de l'acétyl-CoA et ainsi
alimenter son propre cycle de Krebs.
Souvent on oublie en biochimie que ces AA peuvent être aussi des composés qui ont un pouvoir lipogénique
puisque l'Acetyl-CoA est un substrat parfait pour la formation d'AG via le Malonyl-CoA.
L'idée ici c'est de ne pas retenir par coeur le cycle de Krebs mais connaîitre le devenir des AA globalement
dans les différentes situations physiologiques
3) Enzymes clés du métabolisme des acides aminés
D’un point de vue clinique, les 4 enzymes citées ci-dessous ne sont pas les seules mais font partie des plus
importantes, elles sont utilisées comme marqueurs.
- La glutamate déshydrogénase : désamination oxydative, génère de l'alpha-cétoglutarate pour
alimenter le cycle de Krebs qui est favorable pour la néoglucogénèse.
Elle charge le glutamate en ammoniac pour fabriquer de la glutamine. Cette enzyme de désamination
oxydative est importante car si elle n’existait pas, la glutamine ne serait pas fabriquée et ne pourrait
pas transférer l’ammoniac au niveau du foie.
- La glutaminase : fait la réaction inverse. Lorsque la glutamine arrive au niveau du foie et des
reins, il faut qu’elle soit dégradée en glutamate et NH3.
 6
7
8
9
10
11
12
13
14
6
7
8
9
10
11
12
13
14
1
/
14
100%